Class 6: Understanding BLAST
Bioinformatics
Andrés Aravena
November 9, 2023
Looking on a database
We have one sequence, called query
We compare our sequences with each sequence in a Database
(sequences in the database are called subjects)
We get the score of each alignment
We report all subjects with score over a threshold
Columns in tabular output
- qseqid: query sequence id
- sseqid: subject sequence id
- pident: percentage of identical positions
- length: alignment length (sequence overlap)
- mismatch: number of mismatches
- gapopen: number of gap openings
- qstart: start of alignment in query
- qend: end of alignment in query
- sstart: start of alignment in subject
- send: end of alignment in subject
- evalue: expect value
- bitscore: bit score
qseqid and sseqid
query sequence id
subject sequence id
pident and length
percentage of identical positions
- How many letters are the same in the aligned region
alignment length
- May be longer or shorter than the query or the subject
mismatch and gapopen
number of mismatches
- How many letters are different in the aligned region
number of gap openings
- How many initiation of gaps, independent of their length
qstart and qend
start of alignment in query
end of alignment in query
sstart and send
start of alignment in subject
end of alignment in subject
evalue and bitscore
expect value
bit score
Alignment score depends on Substitution matrix
Score can change
If mismatches and gaps have different cost, the score will change
Sometimes the optimal alignment changes
Therefore alignments are meaningless without knowing the scoring matrices
Later we will discuss how to choose the “best” scoring matrix for each case
E-value
What is “a good score”?
We want big scores
How big is big enough?
We need to make several hypothesis
The most common hypothesis is statistical
Larger scores, less hits
A hit is a subject with score over a threshold
Larger score thresholds give less hits
We can estimate the number of hits in a given database, assuming randomness
That is called Expected value
Expected value as a threshold
In practice, we choose a small Expected value
(usually called E-value)
Something like 10-5 or 10-20
What we find is not random
and maybe it is biologically meaningful
E-value depends on the database
The formula for E-value depends on
- The score \(S\)
- The query size \(m\)
- The database size \(n\)
- The substitution scoring matrix, via \(k\) and \(λ\)
\[E=kmn\exp(-λ S)\]
Same alignments in different databases have different E-value
but the same score
Use the smallest relevant database
Many flavors of BLAST
Types of BLAST
Depending on the alphabet of the query and subject
- BlastN
- Search nucleotides in nucleotide databases
- BlastP
- Search proteins in protein databases
- BlastX
- Search nucleotide in protein databases.
- Each query is translated into 6 putative proteins
Types of BLAST
- TBlastN
- Search proteins in nucleotide databases.
- Each subject is translated into 6 putative proteins
- TblastX
- Search nucleotides in nucleotide databases
- Translate each query and each subject into 6 proteins
- Compares all the resulting proteins
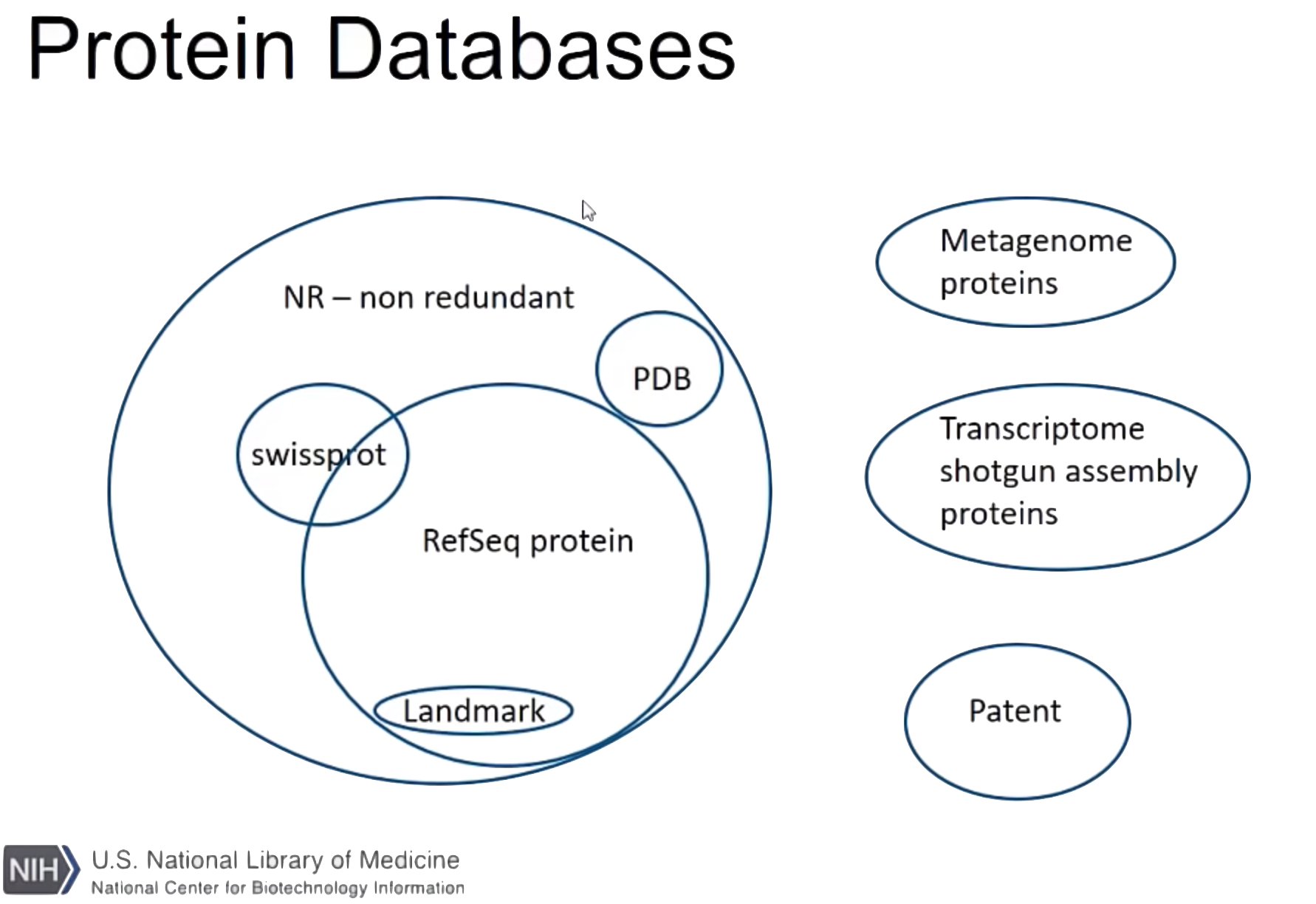
NCBI protein databases
- nr
- Non-redundant protein sequences
- refseq_protein
- Reference proteins
- refseq_select
- Reference Select proteins
What is “Non-Redundant”?
These databases get data from several sources
Sometimes two people upload the same sequence but with different ID
For example, EMBL ID, GenBank ID, RefSeq ID, etc.
This database combines all identical entries into one, and keeps all the alternative IDs
NCBI protein databases
- landmark
- Model Organisms
- swissprot
- UniProtKB/Swiss-Prot
- pat_aa
- Patented protein sequences
NCBI protein databases
- pdb
- Protein Data Bank proteins
- env_nr
- Metagenomic proteins
- tsa_nr
- Transcriptome Shotgun Assembly proteins
NCBI nucleotide databases
- Human G+T
- Human genomic plus transcript
- Mouse G+T
- Mouse genomic plus transcript
- nr/nt
- Nucleotide collection
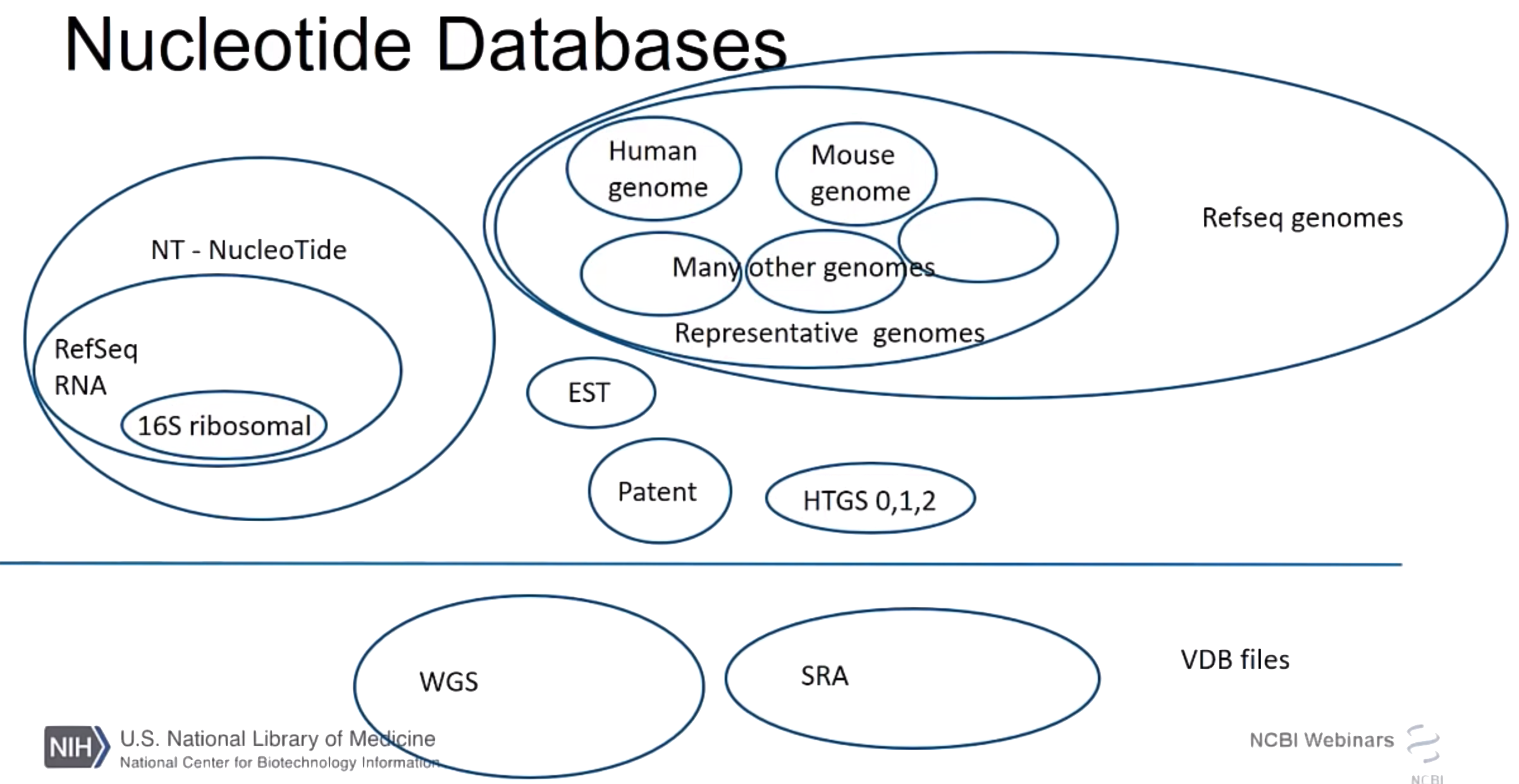
NCBI nucleotide databases
- Bacteria and Archaea
- 16S ribosomal RNA sequences
- refseq_select
- Reference Select sequences
- refseq_rna
- Reference RNA sequences
NCBI nucleotide databases
- refseq_representative_genomes
- RefSeq Representative genomes
- refseq_genomes
- RefSeq Genome Database
NCBI nucleotide (reads)
- SRA
- Sequence Read Archive
- TSA
- Transcriptome Shotgun Assembly
- HTGS
- High throughput genomic sequences
NCBI nucleotide databases
- pat
- Patent sequences
- pdb
- nucleotides in Protein Data Bank
- RefSeq_Gene
- Human RefSeqGene sequences
BlastN variants
- megablast
- Highly similar sequences
- discontiguous megablast
- More dissimilar sequences
- blastn
- Somewhat similar sequences
BlastP variants
- blastp
- protein-protein BLAST.
- PSI-BLAST
- Position-Specific Iterated BLAST.
- builds a position-specific scoring matrix.
- PHI-BLAST
- Pattern Hit Initiated BLAST.
- limits alignments to those that match a pattern in the query.
BlastP variants
- Quick BLASTP
- Accelerated protein-protein BLAST.
- very fast and works best if the target percent identity is 50% or more.
- DELTA-BLAST
- Domain Enhanced Lookup Time Accelerated BLAST.
- builds a PSSM using a Conserved Domain Database search.
- searches a sequence database.