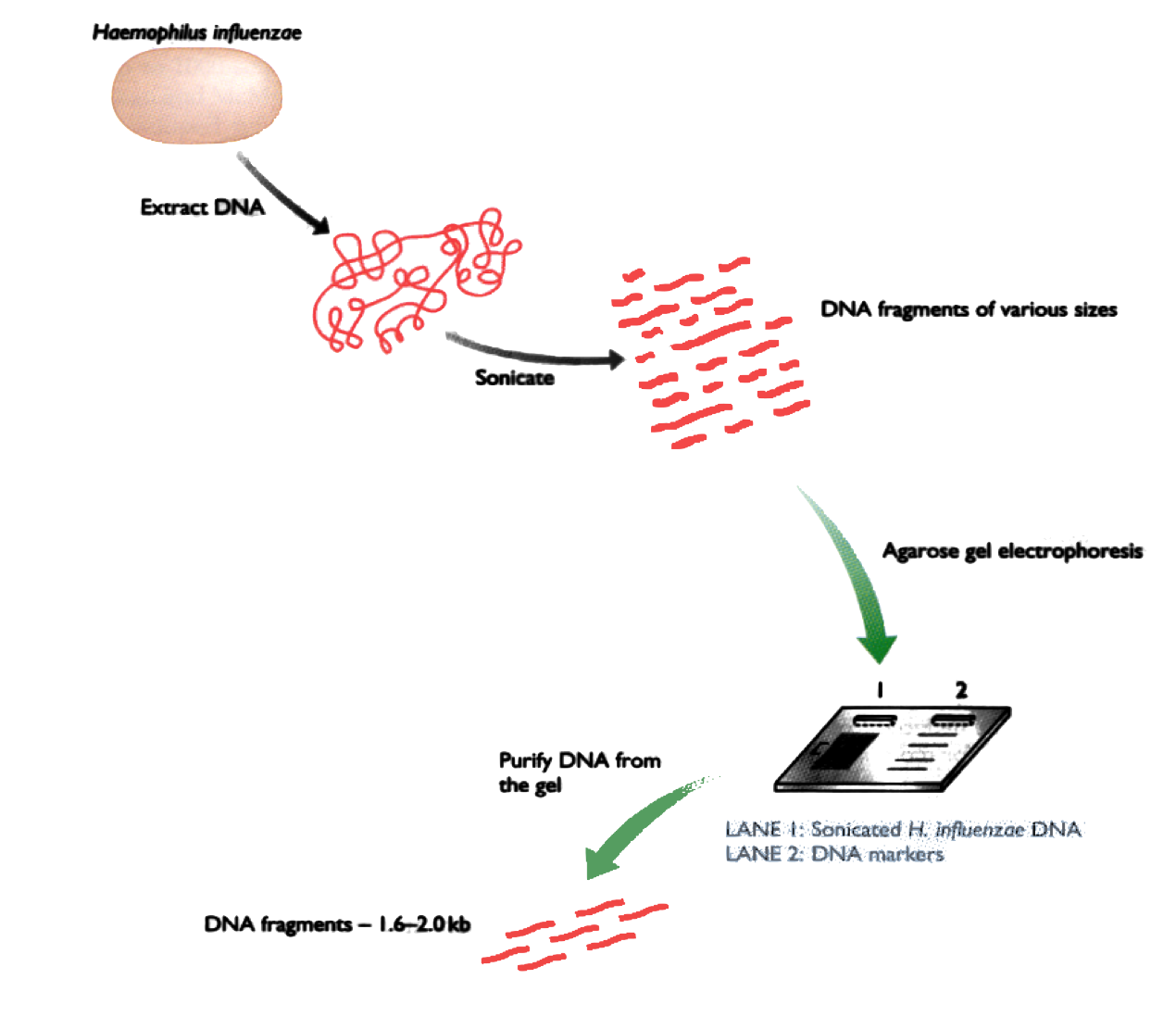
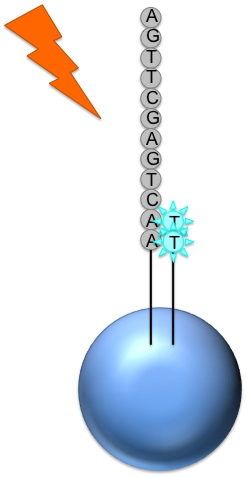
DNA is separated in 4 parts, each part is mixed with a different
restriction enzyme. Then the fragments are separated by
electrophoresis.
In a second version, each part is marked with a different
fluorophores and then placed in a single capilar electrophoresis, with a
light detector
Sanger requires cloning
Cloning vector
know restriction-enzyme cut sites
known binding-sites for primers
antibiotic-resistance genes
replication origin site
Part of the vector will be sequenced in every read
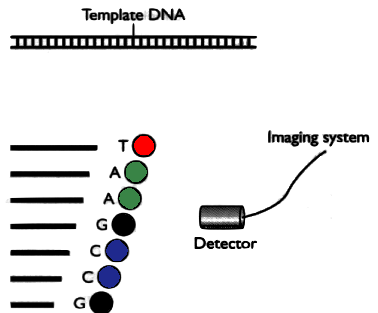
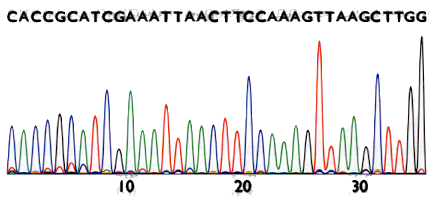
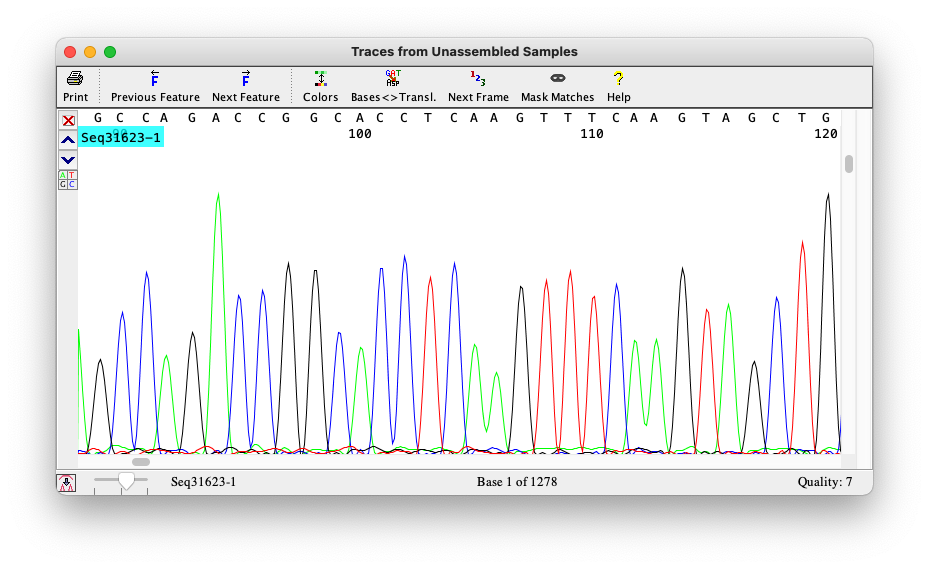
Sanger method’s result
The result is a chromatogram. Filenames ending in
.AB1 or .SCF
New generation sequencing
Roche 454
Illumina Solexa
Ion Torrent
ABI SOLID
Third generation
DNA nanoball
Pacific Biotech (PacBio)
Roche 454
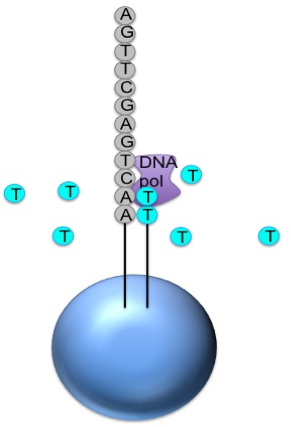
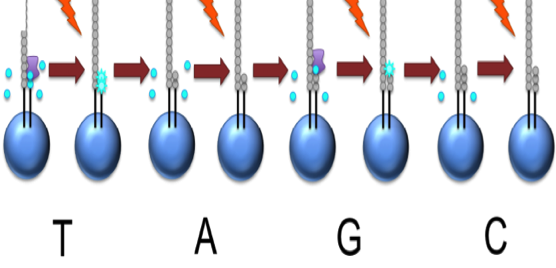
Generic adaptors are added to the ends and annealed to beads, one DNA
fragment per bead
The fragments are then amplified by PCR
Each bead is placed in a single well of a slide
Each well will contain a single bead, covered in many PCR copies of a
single sequence
The slide is flooded with one of the four NTP species
Roche 454
The nucleotide is incorporated when it matches the template
If that single base repeats, then more will be added
The addition of each nucleotide releases a light signal, and is
registered with a hight resolution video
Roche 454
This NTP mix is washed away
The next NTP mix is now added and the process repeated, cycling
through the four NTPs.
This kind of sequencing generates graphs for each sequence read,
showing the signal density for each nucleotide wash
The sequence can then be determined computationally from the signal
density in each wash.
Roche 454
Sequencing by synthesis
can sequence 0.7 gigabase per run
run time 23 hours
read length 700-800 bases
Illumina Solexa
also sequencing by synthesis
old model Genome Analyzer can sequence 1Gb per run
Solexa model can sequence 600 Gb per run
HiSeq model 100 bases read length average 30Gb per run
11 day in regular mode
2 day in rapid run mode
Illumina Solexa
over 70% of the market
≈ $1000 MiSeq
≈ $3000 HighSeq
≈ 1.5 days
Illumina Solexa
output of sequencing data per run: 600 Gb
read lengths: approximately 100 bp
cost is cheap
run times are long: 3-10 days
ABI-SOLID
Sequencing by ligation
Supported Oligonucleotide Ligation and Detection (SOLiD)
Solid 4 model can read 80-100 Gb per run (coverage 30Gb)
average read length 50 bases
fragment length 400-600-bp
ABI-SOLID
The advantage of this method is accuracy with each base interrogated
twice
Major disadvantages
short read lengths (50–75 bp)
very long run times of 7 to 14 days
need for computational infrastructure and expert computing personnel
for analysis of the raw data
Ion torrent
Developed by the inventors of 454 sequencing
With two major changes.
nucleotide sequences are detected electronically by changes in the
pH of the surrounding solution rather than by the generation of
light
sequencing reaction is performed in a microchip with flow cells and
electronic sensors on each cell
The incorporated nucleotide is converted to an electronic
signal
PacBio
PacBio model RS II produces average read lengths over 10 kb, with an
N50 of more than 20 kb and maximum read lengths over 60 kb
Error rate of a continuous long read is relatively high
(around 11%–15%)
The error rate can be reduced by generating circular consensus
sequences reads with sufficient sequencing passes.
Summary
Method
Read length (bp)
Error rate (%)
No. of reads per run
Time per run
Cost per million bases (USD)
Sanger ABI 3730x1
600-1000
0.001
96
0.5–3 h
500
Illumina HiSeq 2500
2 × 250
0.1
1.2 × 109 (pairs)
1–6 days
0.04
PacBio RS II: P6-C4
1.0–1.5×104
13
3.5–7.5 × 104
0.5–4 h
0.4–0.8
Oxford Nanopore MinION
2–5 × 103
38
1.1–4.7 × 104
50 h
6.44–17.9
Base calling
From chromatograms to sequences
Sequencers produce signals
Sanger produces chromatograms
Pyrosequencing make videos
others
each method has their own tool to call the bases
For Sanger, that tool is called Phred
Phred
chromatogram
Phred quality
Signal–Ratio is different for each base
As usual we use logarithms, multiply by a constant, and take the
integer part \[\lfloor10\cdot\log_{10}(\text{Signal}/\text{Noise})\rfloor\]
Phred quality
Q
-Q/10
Prob(Error)
Prob(Correct)
10
-1.0
0.1000000
0.9000000
15
-1.5
0.0316228
0.9683772
20
-2.0
0.0100000
0.9900000
30
-3.0
0.0010000
0.9990000
40
-4.0
0.0001000
0.9999000
50
-5.0
0.0000100
0.9999900
60
-6.0
0.0000010
0.9999990
70
-7.0
0.0000001
0.9999999
80
-8.0
0.0000000
1.0000000
Phred quality of each base
phred quality
Phred file format (.phd)
BEGIN_SEQUENCE GTFAG75TF.scf
BEGIN_COMMENT
CHROMAT_FILE: GTFAG75TF.scf
ABI_THUMBPRINT: 0
TRACE_PROCESSOR_VERSION:
BASECALLER_VERSION: phred 0.071220.c
PHRED_VERSION: 0.071220.c
CALL_METHOD: phred
QUALITY_LEVELS: 99
TIME: Fri Nov 22 15:00:37 2019
TRACE_ARRAY_MIN_INDEX: 0
TRACE_ARRAY_MAX_INDEX: 9116
TRIM: 34 555 0.0500
TRACE_PEAK_AREA_RATIO: 0.0255
CHEM: prim
DYE: rhod
END_COMMENT
BEGIN_DNA
g 6 8
a 6 24
g 6 28
a 8 45
g 8 50
a 8 59
t 8 77
c 8 87
t 9 100
a 9 115
g 11 124
t 11 140
g 15 147
t 8 162
g 9 170
c 12 182
t 9 197
g 17 207
g 20 221
a 21 234
Files of type .qual
This is FASTA format, with numbers instead of letters
Each number is the quality of the corresponding letter