Class 19: Normalization in RNAseq
Systems Biology
Andrés Aravena, PhD
December 28, 2021
Mapping reads to reference genome
There are several tools used to map reads to a genome
bwabowtiebowtie2hisat2
Each one has several options, that you can read in their manual
All of them produces SAM/BAM files
Why they may give different results

Count Normalization
Using featureCounts (or similar) tool, we transform the SAM file into a matrix of counts
The element \(c_{ij}\) is the count of reads for gene \(i\) under library \(j\)
This value depends on
- The gene expression
- The gene length \(l_i\)
- The number of reads in the library \(N_j\)
We care about the first one. The last one depends on technical issues
First idea: correct library effect
We can normalize the counts by the library size
The library size, i.e. the number of reads in the library, is \[N_j = \sum_i c_{ij}\]
So the relative number of reads is \[\frac{c_{ij}}{N_j}\]
These are small numbers
RNAseq libraries are large. They have millions of read
Thus, each \[\frac{c_{ij}}{N_j}\] is a very small number.
We scale them by \(10^6\) and get Reads Per Million \[\frac{c_{ij}⋅10^6}{N_j}\]
(same as we scale \(0.000004m\) to \(4μm\))
Larger genes have more reads
If the reads are taken randomly from each mRNA, then longer genes have higher probability of being sequenced
To correct that we divide by the gene length \(l_i\) \[\frac{c_{ij}⋅10^6}{N_j l_i}\]
Average gene length is around 1000bp
Again, this is a small number, so we scale by \(10^3\) and we get Reads Per Kilo per Million \[RPKM_{ij} = \frac{c_{ij}⋅10^9}{N_j l_i}\]
We can change the division order
It is easy to see that \[RPKM_{ij} = \frac{c_{ij}⋅10^6}{N_j} \frac{10^3}{l_i} = \frac{c_{ij}⋅10^3}{l_i} \frac{10^6}{N_j}\] so we can divide by the gene length first, and the library size later
Fragments instead of Reads
When we use paired-end sequencing, we typically get two reads for each fragment
But sometimes we get only one read
It makes sense to count fragments instead of reads
This is basically handled by the counting step
FPKM is the same formula as RPKM, but counting fragments instead of reads
Comparing libraries
We want to compare at least two libraries
The sum of all RPKM in library \(j_1\) is \[\sum_i RPKM_{i{j_1}} = \frac{10^9}{N_{j_1}}\sum_i \frac{c_{ij}}{l_i}\]
and, in general, this number is different from the sum for another library \(j_2\) \[\sum_i RPKM_{i{j_1}} ≠ \sum_i RPKM_{i{j_2}}\]
That is not good for statistical tests
Another way to normalize
To avoid this issue, we can normalize by the gene length before calculating totals
Let’s call \(Q_i\) to the sum of_gene-length-corrected_counts \[Q_j = \sum_i \frac{c_{ij}}{l_i}\]
Then Transcripts per Million for gene \(i\) in library \(j\) is \[TPM_{ij}=\frac{c_{ij}}{l_i⋅Q_j}\]
Now
Now we have \[\sum_i TPM_{ij}=\frac{1}{Q_j}\sum_i \frac{c_{ij}}{l_i}=1\]
Which one?
RPKM/FPKM are good for comparing between genes in the same library
TPM is better for comparing the same gene between libraries
DESeq2
This tool is based on a different statistical hypothesis
Instead of assuming log-normal distribution, it assumes a negative binomial distribution
Both distributions are similar, but it allows us to see the data from another point of view
In particular, it does not require normalization of counts
DESeq protocol
sw480_APC_1 sw480_APC_2 sw480_APC_3 sw480_CONTROL_1 sw480_CONTROL_2
NM_000014 1 1 3 1 2
NM_000015 1 0 0 0 2
NM_000016 1038 1126 1098 1285 1037
NM_000017 133 111 103 171 136
NM_000018 8699 8232 8364 8888 8426
NM_000019 2094 2070 1879 2645 2248
sw480_CONTROL_3 sw480.1 sw480.2 sw480.3 cds_length gene_name
NM_000014 2 3 2 2 4653 A2M
NM_000015 1 1 3 1 1317 NAT2
NM_000016 1306 1022 1059 1057 2603 ACADM
NM_000017 174 143 147 125 1917 ACADS
NM_000018 9188 10982 10844 11450 2292 ACADVL
NM_000019 2775 2132 2164 2087 2134 ACAT1Describe conditions of each sample
sample_info <- data.frame(
condition=factor(rep(c("APC","CONTROL", "sw480"), each=3),
levels = c("CONTROL", "APC", "sw480")),
row.names = colnames(read.counts[,1:9]))
sample_info condition
sw480_APC_1 APC
sw480_APC_2 APC
sw480_APC_3 APC
sw480_CONTROL_1 CONTROL
sw480_CONTROL_2 CONTROL
sw480_CONTROL_3 CONTROL
sw480.1 sw480
sw480.2 sw480
sw480.3 sw480Pack everything in DESeq format
DESeq.ds <- DESeqDataSetFromMatrix( countData = read.counts[,1:9],
colData = sample_info, design = ~ condition)
colData(DESeq.ds)DataFrame with 9 rows and 1 column
condition
<factor>
sw480_APC_1 APC
sw480_APC_2 APC
sw480_APC_3 APC
sw480_CONTROL_1 CONTROL
sw480_CONTROL_2 CONTROL
sw480_CONTROL_3 CONTROL
sw480.1 sw480
sw480.2 sw480
sw480.3 sw480 DataFrame with 39276 rows and 0 columnsThe data is there
sw480_APC_1 sw480_APC_2 sw480_APC_3 sw480_CONTROL_1 sw480_CONTROL_2
NM_000014 1 1 3 1 2
NM_000015 1 0 0 0 2
NM_000016 1038 1126 1098 1285 1037
NM_000017 133 111 103 171 136
NM_000018 8699 8232 8364 8888 8426
NM_000019 2094 2070 1879 2645 2248
sw480_CONTROL_3 sw480.1 sw480.2 sw480.3
NM_000014 2 3 2 2
NM_000015 1 1 3 1
NM_000016 1306 1022 1059 1057
NM_000017 174 143 147 125
NM_000018 9188 10982 10844 11450
NM_000019 2775 2132 2164 2087Remove genes without any counts
Now we verify that we did not lose any read
[1] TRUENormalization by library
Estimate a factor to correct each library
sw480_APC_1 sw480_APC_2 sw480_APC_3 sw480_CONTROL_1 sw480_CONTROL_2
1.0059487 0.9570850 0.9454599 1.0364710 0.9067061
sw480_CONTROL_3 sw480.1 sw480.2 sw480.3
1.0709679 1.0344438 1.0359450 1.0370205 Normalized counts
It is just a factor of scale
counts.sf_normalized <- counts(DESeq.ds, normalized = TRUE)
plot(counts(DESeq.ds)[,1], counts.sf_normalized[,1], pch=".", log="xy")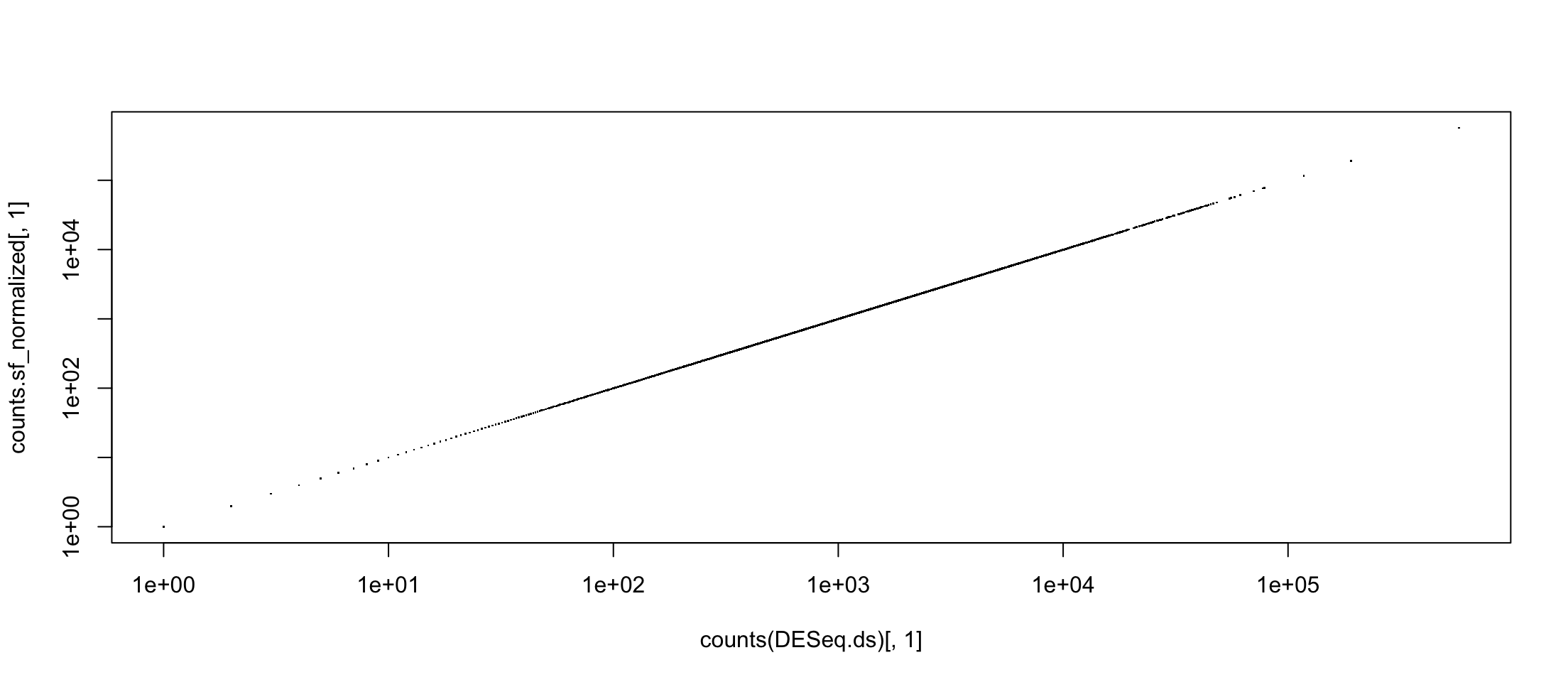
We can use logarithms
Let’s avoid log2(0) by adding a pseudo-count

DESeq does something different
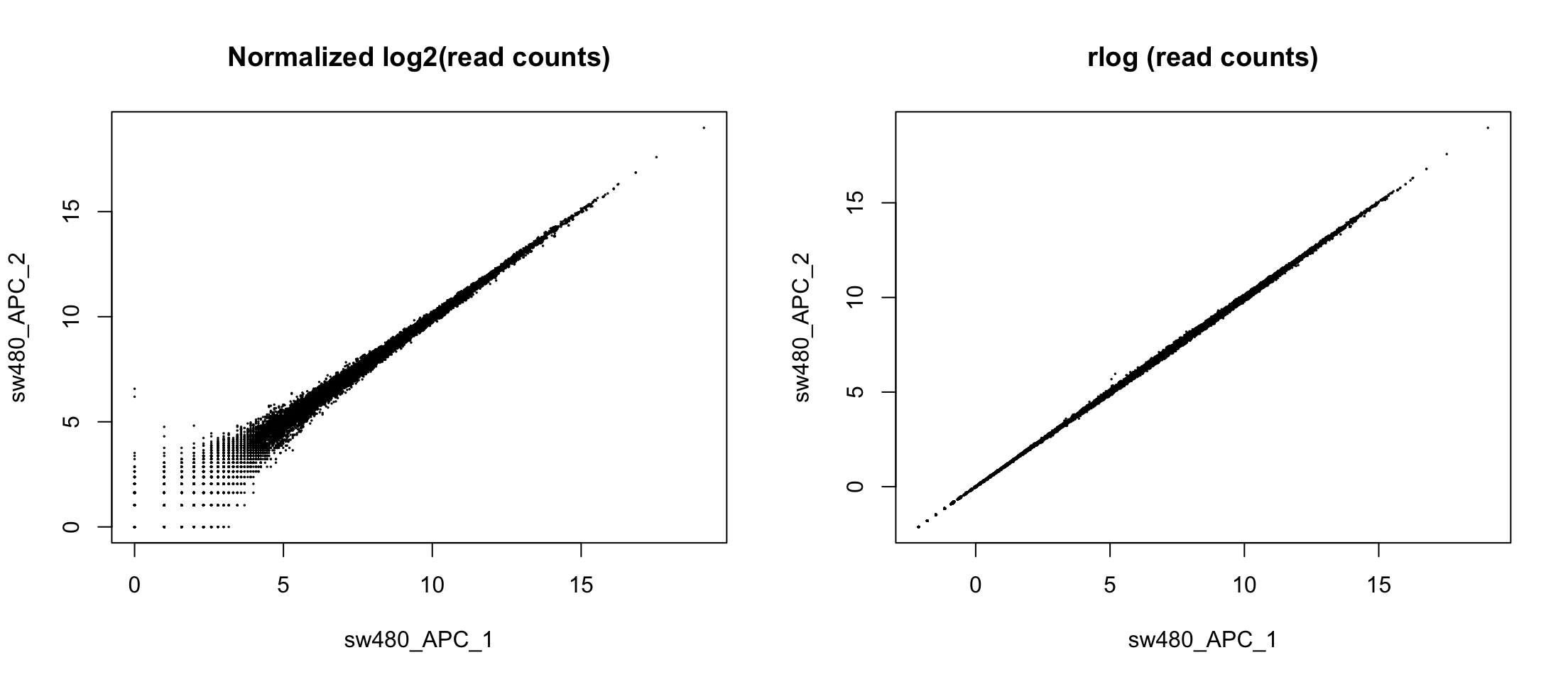
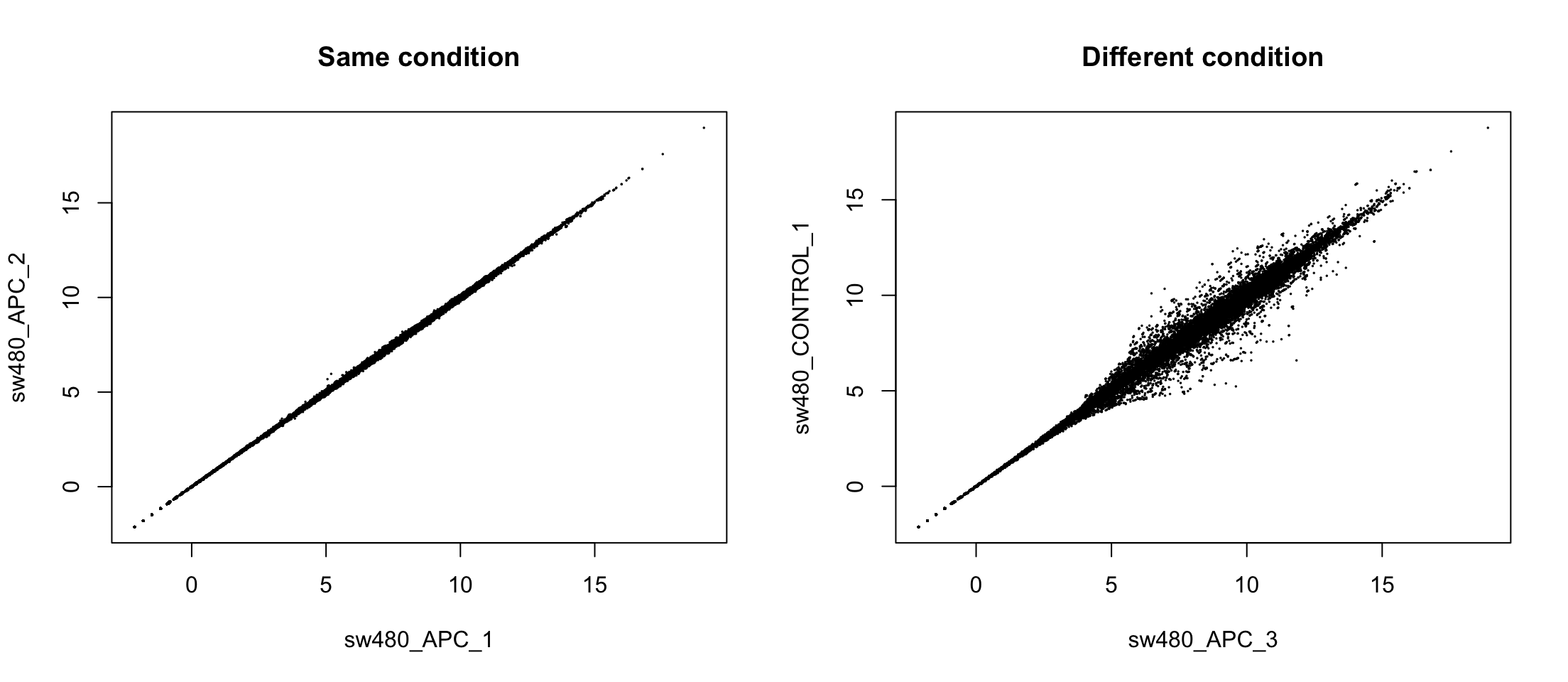
Comparing conditions
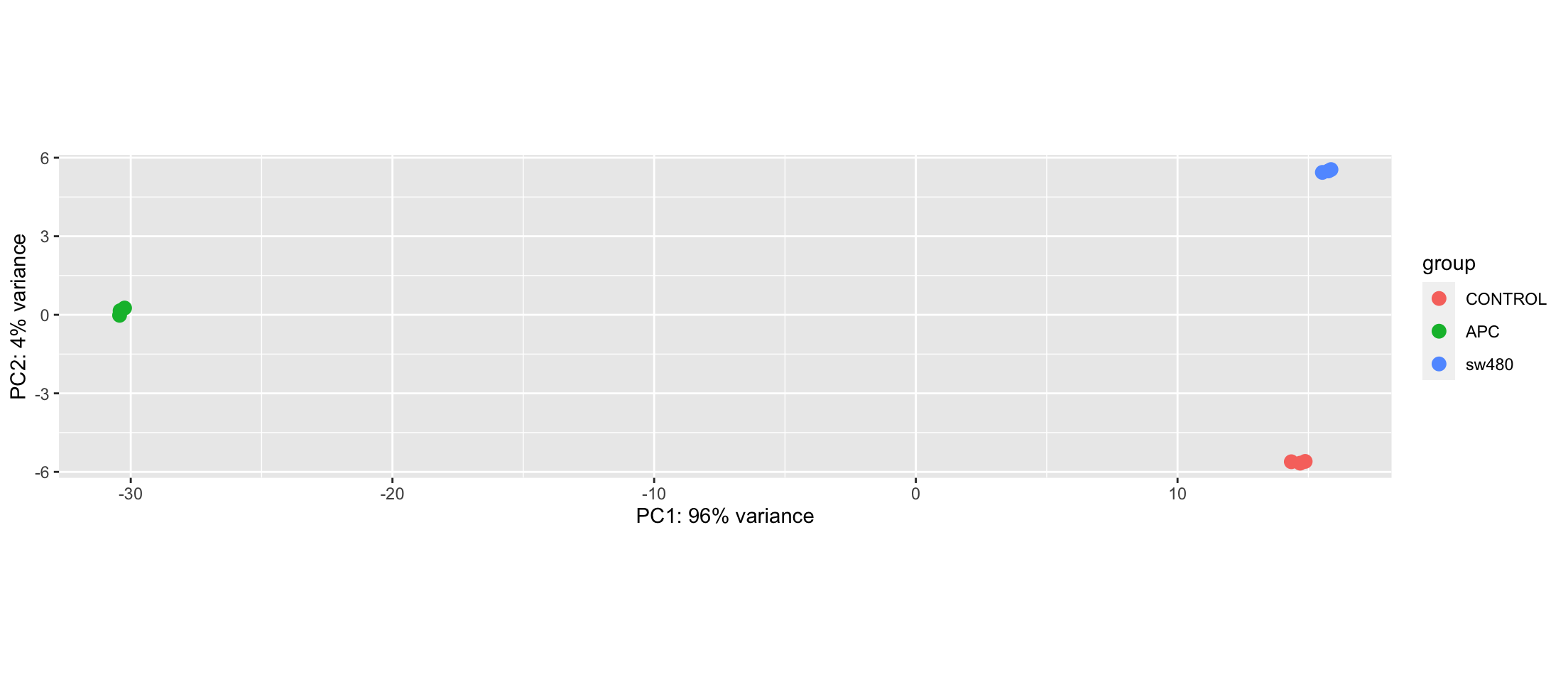
Principal Component Analysis
Let’s do the statistics
First, gene-wise dispersion estimates across all samples
gene-wise dispersion estimatesmean-dispersion relationshipfinal dispersion estimatesThen a t-test (assuming different variances)
Instead of topTable
log2 fold change (MLE): condition sw480 vs CONTROL
Wald test p-value: condition sw480 vs CONTROL
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
NM_000014 1.889930 0.433904 1.1851497 0.366118 7.14277e-01
NM_000015 0.995613 0.655316 1.6196758 0.404597 6.85774e-01
NM_000016 1111.347112 -0.251112 0.0621873 -4.038000 5.39089e-05
NM_000017 137.250467 -0.254684 0.1441359 -1.766968 7.72335e-02
NM_000018 9407.540815 0.281006 0.0384621 7.306038 2.75134e-13
NM_000019 2224.066114 -0.307231 0.0464157 -6.619114 3.61357e-11
padj
<numeric>
NM_000014 NA
NM_000015 NA
NM_000016 2.67474e-04
NM_000017 1.56518e-01
NM_000018 4.40436e-12
NM_000019 4.61581e-10Choosing another condition
results(DESeq.ds, contrast=c("condition", "APC", "CONTROL"),
independentFiltering = TRUE, alpha = 0.05) |> head()log2 fold change (MLE): condition APC vs CONTROL
Wald test p-value: condition APC vs CONTROL
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
NM_000014 1.889930 0.0507329 1.2335629 0.0411271 9.67195e-01
NM_000015 0.995613 -1.4665320 1.8959505 -0.7735076 4.39222e-01
NM_000016 1111.347112 -0.0982781 0.0619814 -1.5856055 1.12829e-01
NM_000017 137.250467 -0.4190118 0.1475355 -2.8400750 4.51029e-03
NM_000018 9407.540815 -0.0189505 0.0387184 -0.4894445 6.24527e-01
NM_000019 2224.066114 -0.2909752 0.0466153 -6.2420551 4.31859e-10
padj
<numeric>
NM_000014 9.83238e-01
NM_000015 5.45796e-01
NM_000016 1.73850e-01
NM_000017 9.33849e-03
NM_000018 7.18658e-01
NM_000019 1.76809e-09Big picture
