Class 11: Clustal
Bioinformatics
Andrés Aravena
November 1st, 2021
Clustal
Clustal was the first popular multiple sequence aligner
Versions: Clustal 1, Clustal 2, Clustal 3, Clustal 4, Clustal V, Clustal W, Clustal X, Clustal Ω
Only the last one is used today
Scoring function
First, there should be a scoring function
Clustal uses a simple one. The sum of all v/s all \[\begin{aligned}
\text{Score}_k(s_{1}[i_1],…,s_{k}[i_k])= & \text{Score}_2(s_{1}[i_1],s_{2}[i_2]) + \\
& \text{Score}_2(s_{1}[i_1],s_{3}[i_3]) + \cdots+ \\
& \text{Score}_2(s_{k-1}[i_{k-1}],s_{k}[i_k])
\end{aligned}\] where \(\text{Score}_2(s_{a}[i],s_{b}[j])\) is PAM, BLOSUM, or a similar substitution scoring matrix
Progressive alignment order
We start by comparing sequences all-to-all
That is, comparing all pairs of sequences
We store them in a distance matrix
How many pairs can be done with \(N\) sequences?
Building a guide tree
Once we get all pairwise “distances”
(that is, scores)
We make a tree by hierarchical clustering
![]()
Hierarchical clustering
Start with one leaf node for each sequence, and no branches
- Take the two sequences (\(A\) and \(B\)) that are more similar (highest score)
- create a new node \(C\) connected to \(A\) and \(B\)
- \(\text{Score}_2(X,C)\) between each old node \(X\) and \(C\) is the average of \(\text{Score}_2(X,A)\) and \(\text{Score}_2(X,B)\) \[\text{Score}_2(X,C)=\frac{\text{Score}_2(X,A)+\text{Score}_2(X,B)}{2}\]
- forget about \(A\) and \(B\)
Hierarchical Clustering
![]()
bottom up: joining one by one
- if \(\mathrm{Score}(x, y)\) is the largest one, we join \(x\) and \(y\)
- we create cluster \(C\)
This is not a phylogenetic tree
The guide tree is built without seeing the big picture
So it is not safe to assign any meaning to it
We will talk more about trees and build phylogenetic trees later
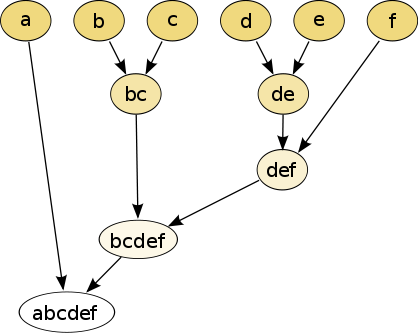
Guide tree guides the alignment
![]()
Clustal aligns the sequences following the guide tree
First, it aligns the more similar sequences
Then it adds the nearest sequence, and so on
These are semi-global alignments
Uses \(\text{Score}_k()\) when there are \(k\) sequences