
- A big molecule, but not too complex. It is a polymer
- All made of only 4 pieces (so there is a pattern)
March 10, 2020

Despite being large molecules, DNA and proteins are made with pieces of only a few types
DNA is made of four bases. We can represent it with four letters
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGC
Proteins have 20 aminoacids. We can represent them with 20 letters.
MRVLKFGGTSVANAERFLRVADILESNARQGQVATVLSAPAKITNHLVAMIEKTISGQDALPNISDAERI
In molecular biology we often work with sequences
The main reason why computing is useful for molecular biology
ATGAATACTATATTTTCAAGAATAACACCATTAGGAAATGGTACGTTATGTGTTATAAGAATTTCTGGAA AAAATGTAAAATTTTTAATACAAAAAATTGTAAAAAAAAATATAAAAGAAAAAATAGCTACTTTTTCTAA ATTATTTTTAGATAAAGAATGTGTAGATTATGCAATGATTATTTTTTTTAAAAAACCAAATACGTTCACT GGAGAAGATATAATCGAATTTCATATTCACAATAATGAAACTATTGTAAAAAAAATAATTAATTATTTAT TATTAAATAAAGCAAGATTTGCAAAAGCTGGCGAATTTTTAGAAAGACGATATTTAAATGGAAAAATTTC TTTAATAGAATGCGAATTAATAAATAATAAAATTTTATATGATAATGAAAATATGTTTCAATTAACAAAA AATTCTGAAAAAAAAATATTTTTATGTATAATTAAAAATTTAAAATTTAAAATAAATTCTTTAATAATTT GTATTGAAATCGCAAATTTTAATTTTAGTTTTTTTTTTTTTAATGATTTTTTATTTATAAAATATACATT TAAAAAACTATTAAAACTTTTAAAAATATTAATTGATAAAATAACTGTTATAAATTATTTAAAAAAGAAT TTCACAATAATGATATTAGGTAGAAGAAATGTAGGAAAGTCTACTTTATTTAATAAAATATGTGCACAAT ATGACTCGATTGTAACTAATATTCCTGGTACTACAAAAAATATTATATCAAAAAAAATAAAAATTTTATC TAAAAAAATAAAAATGATGGATACAGCAGGATTAAAAATTAGAACTAAAAATTTAATTGAAAAAATTGGA ATTATTAAAAATATAAATAAAATTTATCAAGGAAATTTAATTTTGTATATGATTGATAAATTTAATATTA AAAATATATTTTTTAACATTCCAATAGATTTTATTGATAAAATTAAATTAAATGAATTAATAATTTTAGT TAACAAATCAGATATTTTAGGAAAAGAAGAAGGAGTTTTTAAAATAAAAAATATATTAATAATTTTAATT TCTTCTAAAAATGGAACTTTTATAAAAAATTTAAAATGTTTTATTAATAAAATCGTTGATAATAAAGATT TTTCTAAAAATAATTATTCTGATGTTAAAATTCTATTTAATAAATTTTCTTTTTTTTATAAAGAATTTTC ATGTAACTATGATTTAGTGTTATCAAAATTAATTGATTTTCAAAAAAATATATTTAAATTAACAGGAAAT TTTACTAATAAAAAAATAATAAATTCTTGTTTTAGAAATTTTTGTATTGGTAAATGAATATTTTTAATAT AATTATTATTGGAGCAGGACATTCTGGTATAGAAGCAGCTATATCTGCATCTAAAATATGTAATAAAATA
There are several ways to store DNA or protein data
Most of the times they are stored in FASTA format
FASTA files are text files, with some rules
Microsoft Word files (doc or docx) are NOT text files
You should never use Microsoft Word to store sequences
>AP009180.1 Candidatus Carsonella ruddii PV DNA, complete genome ATGAATACTATATTTTCAAGAATAACACCATTAGGAAATGGTACGTTATGTGTTATAAGAATTTCTGGAA AAAATGTAAAATTTTTAATACAAAAAATTGTAAAAAAAAATATAAAAGAAAAAATAGCTACTTTTTCTAA ATTATTTTTAGATAAAGAATGTGTAGATTATGCAATGATTATTTTTTTTAAAAAACCAAATACGTTCACT GGAGAAGATATAATCGAATTTCATATTCACAATAATGAAACTATTGTAAAAAAAATAATTAATTATTTAT TATTAAATAAAGCAAGATTTGCAAAAGCTGGCGAATTTTTAGAAAGACGATATTTAAATGGAAAAATTTC TTTAATAGAATGCGAATTAATAAATAATAAAATTTTATATGATAATGAAAATATGTTTCAATTAACAAAA AATTCTGAAAAAAAAATATTTTTATGTATAATTAAAAATTTAAAATTTAAAATAAATTCTTTAATAATTT GTATTGAAATCGCAAATTTTAATTTTAGTTTTTTTTTTTTTAATGATTTTTTATTTATAAAATATACATT TAAAAAACTATTAAAACTTTTAAAAATATTAATTGATAAAATAACTGTTATAAATTATTTAAAAAAGAAT TTCACAATAATGATATTAGGTAGAAGAAATGTAGGAAAGTCTACTTTATTTAATAAAATATGTGCACAAT ATGACTCGATTGTAACTAATATTCCTGGTACTACAAAAAATATTATATCAAAAAAAATAAAAATTTTATC TAAAAAAATAAAAATGATGGATACAGCAGGATTAAAAATTAGAACTAAAAATTTAATTGAAAAAATTGGA ATTATTAAAAATATAAATAAAATTTATCAAGGAAATTTAATTTTGTATATGATTGATAAATTTAATATTA AAAATATATTTTTTAACATTCCAATAGATTTTATTGATAAAATTAAATTAAATGAATTAATAATTTTAGT TAACAAATCAGATATTTTAGGAAAAGAAGAAGGAGTTTTTAAAATAAAAAATATATTAATAATTTTAATT TCTTCTAAAAATGGAACTTTTATAAAAAATTTAAAATGTTTTATTAATAAAATCGTTGATAATAAAGATT TTTCTAAAAATAATTATTCTGATGTTAAAATTCTATTTAATAAATTTTCTTTTTTTTATAAAGAATTTTC ATGTAACTATGATTTAGTGTTATCAAAATTAATTGATTTTCAAAAAAATATATTTAAATTAACAGGAAAT TTTACTAATAAAAAAATAATAAATTCTTGTTTTAGAAATTTTTGTATTGGTAAATGAATATTTTTAATAT AATTATTATTGGAGCAGGACATTCTGGTATAGAAGCAGCTATATCTGCATCTAAAATATGTAATAAAATA
… and more
>NC_000913.3_prot_NP_414542.1_1 [gene=thrL] [protein=thr operon leader peptide] [protein_id=NP_414542.1] [location=190..255] MKRISTTITTTITITTGNGAG >NC_000913.3_prot_NP_414543.1_2 [gene=thrA] [protein=Bifunctional aspartokinase/homoserine dehydrogenase 1] [protein_id=NP_414543.1] [location=337..2799] MRVLKFGGTSVANAERFLRVADILESNARQGQVATVLSAPAKITNHLVAMIEKTISGQDALPNISDAERI FAELLTGLAAAQPGFPLAQLKTFVDQEFAQIKHVLHGISLLGQCPDSINAALICRGEKMSIAIMAGVLEA RGHNVTVIDPVEKLLAVGHYLESTVDIAESTRRIAASRIPADHMVLMAGFTAGNEKGELVVLGRNGSDYS AAVLAACLRADCCEIWTDVDGVYTCDPRQVPDARLLKSMSYQEAMELSYFGAKVLHPRTITPIAQFQIPC LIKNTGNPQAPGTLIGASRDEDELPVKGISNLNNMAMFSVSGPGMKGMVGMAARVFAAMSRARISVVLIT QSSSEYSISFCVPQSDCVRAERAMQEEFYLELKEGLLEPLAVTERLAIISVVGDGMRTLRGISAKFFAAL ARANINIVAIAQGSSERSISVVVNNDDATTGVRVTHQMLFNTDQVIEVFVIGVGGVGGALLEQLKRQQSW LKNKHIDLRVCGVANSKALLTNVHGLNLENWQEELAQAKEPFNLGRLIRLVKEYHLLNPVIVDCTSSQAV ADQYADFLREGFHVVTPNKKANTSSMDYYHQLRYAAEKSRRKFLYDTNVGAGLPVIENLQNLLNAGDELM KFSGILSGSLSYIFGKLDEGMSFSEATTLAREMGYTEPDPRDDLSGMDVARKLLILARETGRELELADIE IEPVLPAEFNAEGDVAAFMANLSQLDDLFAARVAKARDEGKVLRYVGNIDEDGVCRVKIAEVDGNDPLFK VKNGENALAFYSHYYQPLPLVLRGYGAGNDVTAAGVFADLLRTLSWKLGV >NC_000913.3_prot_NP_414544.1_3 [gene=thrB] [protein=homoserine kinase] [protein_id=NP_414544.1] [location=2801..3733] MVKVYAPASSANMSVGFDVLGAAVTPVDGALLGDVVTVEAAETFSLNNLGRFADKLPSEPRENIVYQCWE RFCQELGKQIPVAMTLEKNMPIGSGLGSSACSVVAALMAMNEHCGKPLNDTRLLALMGELEGRISGSIHY DNVAPCFLGGMQLMIEENDIISQQVPGFDEWLWVLAYPGIKVSTAEARAILPAQYRRQDCIAHGRHLAGF IHACYSRQPELAAKLMKDVIAEPYRERLLPGFRQARQAVAEIGAVASGISGSGPTLFALCDKPETAQRVA DWLGKNYLQNQEGFVHICRLDTAGARVLEN >NC_000913.3_prot_NP_414545.1_4 [gene=thrC] [protein=L-threonine synthase] [protein_id=NP_414545.1] [location=3734..5020] MKLYNLKDHNEQVSFAQAVTQGLGKNQGLFFPHDLPEFSLTEIDEMLKLDFVTRSAKILSAFIGDEIPQE ILEERVRAAFAFPAPVANVESDVGCLELFHGPTLAFKDFGGRFMAQMLTHIAGDKPVTILTATSGDTGAA VAHAFYGLPNVKVVILYPRGKISPLQEKLFCTLGGNIETVAIDGDFDACQALVKQAFDDEELKVALGLNS ANSINISRLLAQICYYFEAVAQLPQETRNQLVVSVPSGNFGDLTAGLLAKSLGLPVKRFIAATNVNDTVP RFLHDGQWSPKATQATLSNAMDVSQPNNWPRVEELFRRKIWQLKELGYAAVDDETTQQTMRELKELGYTS EPHAAVAYRALRDQLNPGEYGLFLGTAHPAKFKESVEAILGETLDLPKELAERADLPLLSHNLPADFAAL RKLMMNHQ >NC_000913.3_prot_NP_414546.1_5 [gene=yaaX] [protein=DUF2502 family putative periplasmic protein] [protein_id=NP_414546.1] [location=5234..5530] MKKMQSIVLALSLVLVAPMAAQAAEITLVPSVKLQIGDRDNRGYYWDGGHWRDHGWWKQHYEWRGNRWHL HGPPPPPRHHKKAPHDHHGGHGPGKHHR >NC_000913.3_prot_NP_414547.1_6 [gene=yaaA] [protein=peroxide resistance protein, lowers intracellular iron] [protein_id=NP_414547.1] [location=complement(5683..6459)] MLILISPAKTLDYQSPLTTTRYTLPELLDNSQQLIHEARKLTPPQISTLMRISDKLAGINAARFHDWQPD FTPANARQAILAFKGDVYTGLQAETFSEDDFDFAQQHLRMLSGLYGVLRPLDLMQPYRLEMGIRLENARG
We use this genome in classes because it is a small example
Candidatus Carsonella ruddii is a obligate symbiont of Pachpsylla venusta.
There is much controversy over whether this is a living cell or simply an organelle as it is missing genes needed for living independently.
Published as “The 160-kilobase genome of the bacterial endosymbiont Carsonella.” https://www.ncbi.nlm.nih.gov/pubmed/17038615
NCBI stores all public biological sequences at
https://www.ncbi.nlm.nih.gov/nuccore
Anybody can upload sequences, and they may be wrong
NCBI has a curation process to validate the sequences
If a sequence is good enough to be a reference, then it is stored in the RefSeq collection https://www.ncbi.nlm.nih.gov/refseq
Accession numbers are the best way to identify a biological sequence
Different sequences can have the same name, but never the same accession
NCBI search box looks for patterns everywhere, not only on the title
and many sequences can have the same name
To be sure of using the correct sequence, go to NCBI Taxonomy
https://www.ncbi.nlm.nih.gov/taxonomy
Try with “Escherichia coli”. There are many
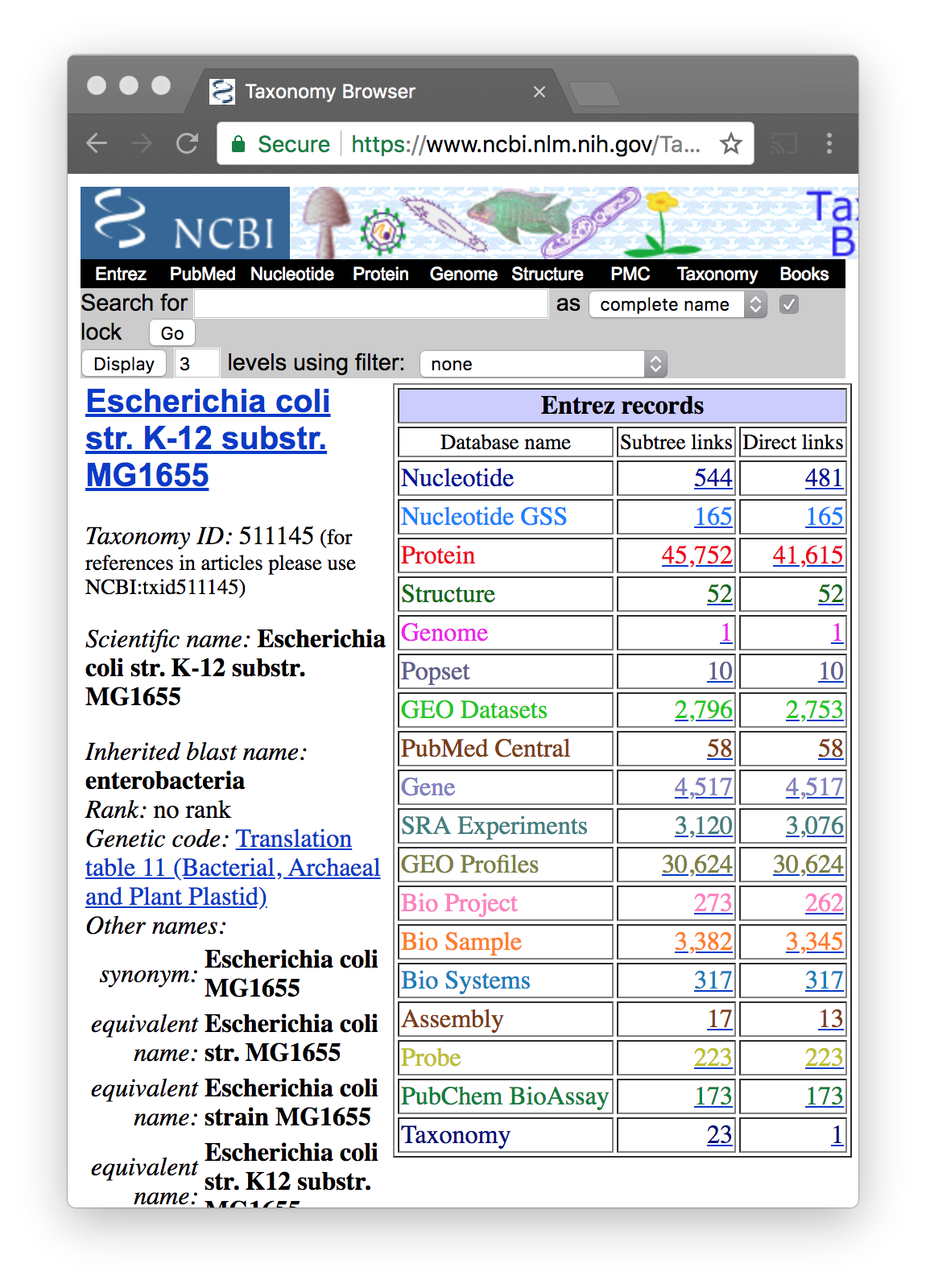
You will see that each organism has a taxonomic ID
You click on Nucleotide–Direct Links
In the search box add the phrase “complete genome”
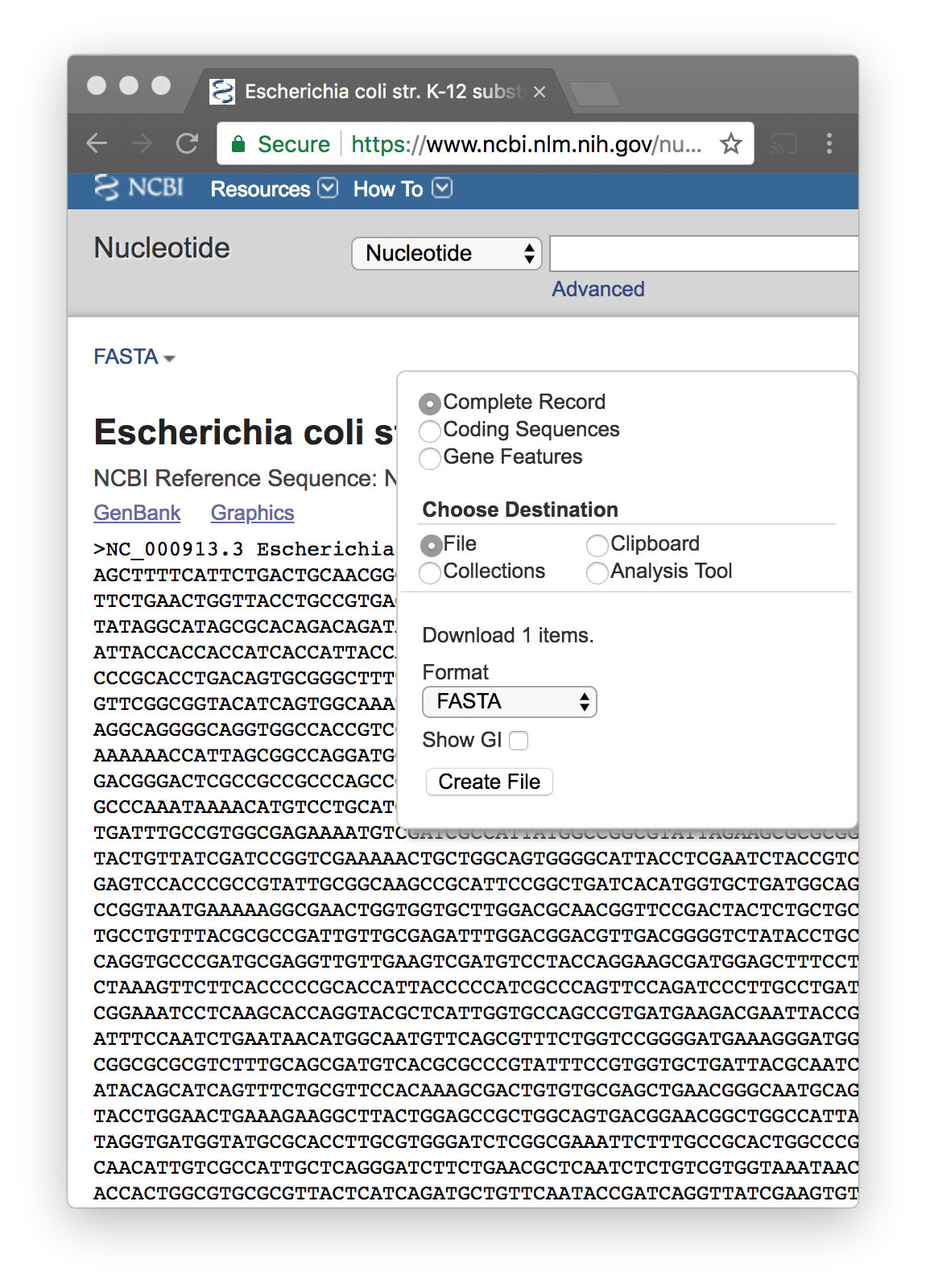
You download the FASTA file
Store it on your computer, and change the name
If you use the Lab’s computers, save it on X:
To handle sequence data in R, we use the seqinr library
You have to install it once.
install.packages("seqinr")
Then you have to load it on every session
library(seqinr)
read.fasta(file, seqtype = "DNA", ...)
A list of vectors of chars. Each element is a sequence object.
library(seqinr)
sequences <- read.fasta("NC_000913.faa")
This is a list of protein sequences
The first sequence is sequences[[1]]
read.fasta returns a list of vectors of charactersStatistics is a way to tell a story that makes sense of the data
In genomics, we look for biological sense
That story can be global: about the complete genome
Or can be local: about some region of the genome
We will start with global properties
The percentage of nitrogenous bases on a DNA molecule that are either guanine or cytosine.
Measuring the melting temperature of the DNA double helix using spectrophotometry
If the DNA has been sequenced then the GC-content can be accurately calculated by simple arithmetic.
GC-content percentage is calculated as \[\frac{G+C}{A+T+G+C}\]
Write the code for this function:
genome_gc_content <- function(genome) {
# count letters
return(answer)
}
Then use it to find the GC content of E.coli
Is the GC content uniform through all genome?
Do all genes have the same GC content?
What function do we need to answer this question?
What is the name, the inputs, and the output?
Write the code for this function:
genes_gc_content <- function(genes) {
# calculate GC content of each gene
return(answer)
}
What is the data structure of answer?
Calculate the GC content for only part of the genome?
Instead of all the genome, we only look through a window
The result should depend on:
The new function looks like this
gc_content <- function(sequence, position, size) {
# code here
return(answer)
}
What should be the code?
gc_content() in many placesWe want to evaluate gc_content on different positions of the genome
positions <- seq(from=1, to=length(genome), by= window_size)
Now we apply gc_content to each element of positions?
for(i in 1:length(positions)) {
gc[i] <- gc_content(genome, positions[i], window_size)
}