Class 16: Gene expression analysis
Bioinformatics
Andrés Aravena
January 22, 2021
Measuring Gene Expression
More precisely, mRNA concentration
What is the question?
We want to know
- Which genes are being expressed
- How much of each gene is being expressed
- How does expression change
- In time
- Under different conditions
- Between strains/mutants/cell lines
The Big Assumption
Measuring protein concentration is hard
We assume that protein concentration is proportional to mRNA concentration
- Which genes are being transcribed
- How much of each gene is being transcribed
- How does transcription change
- In time
- Under different conditions
- Between strains/mutants/cell lines
qPCR
If you have primers for each gene
- specific to each gene
- thermodynamically stable
- efficient
Raw data: CT value for each gene/condition
and CT value for calibration reference
Hybridization methods
Southern/Northern/Western blot can detect, but not quantify
(I think so. I’m not a biologist)
Instead, we have macro- and microarrays
Raw data: Light intensity (luminescence) in one or more wave length
This is measured in arbitrary units, and is a number between 0 and 65536
(that is, a 16-bits value)
Problem: it is hard to compare genes
- Light intensity depends on hybridization rate
- Hybridization depends on oligo thermodynamics
- GC content can have a big effect
- secondary structure blocks hybridization
Solution
Compare each gene with itself in a different condition
In other words, evaluate differential expression
- we cannot compare gene expression between genes
- we can compare differential gene expression between genes
RNAseq
mRNA is retro-transcribed and fragmented.
Fragments are sequenced. Reads are aligned to reference genome
Raw data: SAM/BAM file with location of each read in the reference genome
Processed data: Number of reads per gene
Problem: it is hard to compare genes
- longer genes get more reads
- different conditions get different number of reads
Solution 1: Normalization
- Divide by library size
- Divide by gene size
Normalization: RPKM, FPKM
Reads per kilobase per million:
- Count reads in a sample
- divide that number by 1E6 (“per million”)
- Divide the read counts by the “per million” scaling factor
- This normalizes for sequencing depth
- We get reads per million (RPM)
- Divide the RPM values by the length of the gene, in kilobases
- This gives you RPKM.
FPKM is the same, but with fragments instead of reads
Normalization: TPM
Transcripts per million: similar to RPKM and FPKM
Just a different order of operations
- Divide the read counts by the length of each gene in kilobases
- This gives reads per kilobase (RPK)
- Count up all the RPK values in a sample and divide this number by 1E6
- This is “per million” scaling factor.
- Divide the RPK values by the “per million” scaling factor
- This gives TPM
Solution 2
Compare each gene with itself in a different condition
In other words, evaluate differential expression
- we cannot compare gene expression between genes
- we can compare differential gene expression between genes
Differential Expression
Let’s say we have two conditions:
- wild-type cells
- mutant cells
Let’s take wild type as the base condition.
Fold change
For a given gene G, we want to calculate \[\frac{\text{Expression}(M)}{\text{Expression}(WT)}\]
This would be a number between 0 and ∞
When the gene is under-expressed, we get something between 0 and 1
When it is over-expressed, we get something between 1 and ∞
Log fold change
It is better to have a symmetrical result \[\log_2 \left(\frac{\text{Expression}(M)}{\text{Expression}(WT)}\right)\]
We use base 2 to get fold-change
- Positive numbers indicate over-expression
- Negative numbers indicate under-expression
- Zero indicates no-change
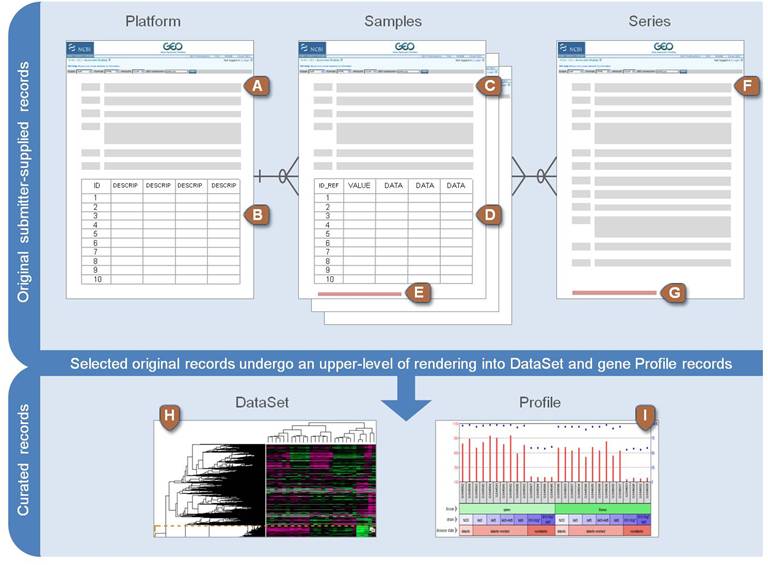
Data source: NCBI GEO
Gene Expression Omnibus
- Platforms
- Samples
- Series
- Data Set
- Profile
NCBI GEO data structure
Application: gene expression analysis
Let’s analyze gene_expression.csv
Each column is a gene, each row is a sample, taken from NCBI GSE95670. The values are absolute expression, we want to know
- The fold-change in expression
- If that change is real, or just noise
Factors indicate the condition
- The first three rows are mutants
- The last three rows are wild-type, or control
We can add a column with a factor describing the condition
Now we can do a model for each gene
We want to measure fold-change. That means, we want to calculate log_2_(mutant/wild type)
Call:
lm(formula = log2(LOC100652730) ~ condition, data = m)
Coefficients:
(Intercept) conditionMutant
7.2767 0.2465 Statistical analysis
Call:
lm(formula = log2(LOC100652730) ~ condition, data = m)
Residuals:
1 2 3 4 5 6
0.5004 -0.2829 -0.2175 0.3705 -0.2239 -0.1466
Coefficients:
Estimate Std. Error t value Pr(>|t|)
(Intercept) 7.2767 0.2211 32.912 5.08e-06 ***
conditionMutant 0.2465 0.3127 0.788 0.475
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Residual standard error: 0.383 on 4 degrees of freedom
Multiple R-squared: 0.1344, Adjusted R-squared: -0.08195
F-statistic: 0.6213 on 1 and 4 DF, p-value: 0.4747Confidence intervals
2.5 % 97.5 %
(Intercept) 6.6628636 7.890598
conditionMutant -0.6216828 1.114595Check if the confidence interval contains 0
Other cases
2.5 % 97.5 %
conditionMutant 0.5694141 3.608096 2.5 % 97.5 %
conditionMutant -1.433264 -0.2166465 2.5 % 97.5 %
conditionMutant 4.45331 8.712368 2.5 % 97.5 %
conditionMutant -4.362937 -2.184466