Class 10: Phylogenetic Trees
Bioinformatics
Andrés Aravena
4 December 2020
we know sequences today, we want to know how they come to be
Let’s think about bacteria
If an organism X evolves into two new organisms A and B, both new organisms share something in common
For example
X: TGGGGCAAGTCGGATCCAGATGGGCGCTAC
A: TGGGGCAAGTCGGATCCAGATGGGCGCTAT
B: TAGGGCAAGTCGGATCCAGATGGGCGCTACIf we had a time machine…
We would see evolution like this

But we do not have one
so we only see the modern organisms

The question is
How to reconstruct the original tree, given the modern sequences
How evolution works
See it working
How evolution works
- random mutations
- selection
- competition for the environment: bottleneck
- coevolution
Random mutations
DNA replication is not 100% perfect
Mutations can be
- substitutions
- insertions
- deletions
- reorganizations
Selection
- not all mutations are “accepted”
- probably most mutations are lethal
- we only see mutations that keeps the organism alive
- Some mutations can give an advantage
- Other mutations are neutral
Competition
- in the short term, all viable organisms are alive
- in the long term, and when resources are scarce, some organisms do not survive
- for example, some organisms may be more efficient in capturing food or using energy
- some organisms have higher “fitness”
- if the environment changes, the “fitness” changes
- there may be bottleneck effects
Coevolution
- Evolution is more complex for sexual organisms
- some individuals do not pass their genes to the next generation, due to mate-selection
- mate-selection also evolves
- We say that phenotype and peer-selection co-evolve
Coevolution also between predator & prey
“Every morning in Africa, a gazelle wakes up, it knows it must run faster than the fastest lion or it will be killed.
“Every morning in Africa, a lion wakes up, it knows it must run faster than the slowest gazelle, or it will starve.
“It doesn’t matter whether you’re the lion or a gazelle-when the sun comes up, you’d better be running.”
Molecular evolution
Looking at only one gene
For this class we will consider the 16S gene in bacteria
- Approx. 1500 nucleotides
- Highly conserved
- Most mutations are lethal
- Cell viability depends on 16S structure
- Asexual reproduction
Trees are networks
Trees are a kind of networks where every node is connected and there are no loops
There are two kinds of nodes
- internal nodes
- they have “children”
- leaves
- They do not have “children”
We see only the leaves, we want to find the internal nodes, and the branches
Rooted trees
To represent evolution we start with an initial node
It is the ancestor to all other nodes, and it has no ancestor
It has two arrows pointing to its “children”
The arrows start from the root and point to the leaves.
Unrooted trees
Looking only at the modern data, we cannot know which sequence existed before
That is, we cannot put an arrow between two nodes
We put a link, undirected, between nodes
These trees are called unrooted
Outgroups point to the root
Since we only see leaves, we cannot put arrows
So we cannot tell which internal node is the root
But, if we include a leave that we know is very distant from all the others, then we can find the root.
Illustration: Unrooted tree

Illustration: Unrooted tree with outgroup

Illustration: Rooted tree

Mathematical Language
At least two groups of people have worked with networks: engineers and mathematicians
They use different words for the same objects
Network: graph
Node: vertice
Link: edge
Arrow: arc
Essence of a tree
The same tree can be drawn in several ways
The drawing is not important
The only important things are
- The tree topology. That is, who is connected to who
- The length of each arc (or edge)
Reconstructing the tree
there are basically three approaches
- Maximum parsimony
- smallest tree that explains all mutations
- Maximum likelihood
- most probable tree, using a probabilistic model
- Distance based
- forget the sequences, use only their distances
In all cases the input is a multiple alignment of all sequences
Maximum parsimony
If we know the tree topology, we can count how many mutations are needed to match our data

So we just have to test all trees and see which one is the best
There are too many trees
But the number of trees is HUGE (\(n^{n-2}\))
So the search has to be done with heuristics
Other problem with parsimony methods
- In some simulations the predicted tree may be very different from the real one
- We only know “the real tree” when we create it
- It can be statistically inconsistent
- That is, adding more sequences sometimes makes a worse tree
Maximum likelihood
An alternative is to find the most probable tree, given the available data
This method needs:
- A probabilistic model of evolution
- Looking at all the trees
So, again, we need an heuristic
Distance methods
Here we use the multiple alignment to calculate the distance between sequences. For example
| a | b | c | d | e | |
|---|---|---|---|---|---|
| a | 0 | 17 | 21 | 31 | 23 |
| b | 17 | 0 | 30 | 34 | 21 |
| c | 21 | 30 | 0 | 28 | 39 |
| d | 31 | 34 | 28 | 0 | 43 |
| e | 23 | 21 | 39 | 43 | 0 |
We call it \(D_1.\) Then we forget about the sequences
Example data from Wikipedia
Hierarchical clustering WPGMA
The smallest distance in \(D_1\) is \(D_1 (a,b)=17\)
We join \(a\) and \(b\) into a new node \((a,b)\), and update distances
Updated distances
\[ \begin{aligned} D_2((a,b),c)= & \frac{D_1(a,c) + D_1(b,c)}{2}=\frac{21+30}{2}=25.5\\ D_2((a,b),d)= & \frac{D_1(a,d) + D_1(b,d)}{2}=\frac{31+34}{2}=32.5\\ D_2((a,b),e)= & \frac{D_1(a,e) + D_1(b,e)}{2}=\frac{23+21}{2}=22 \end{aligned} \]
Updated matrix
| (a,b) | c | d | e | |
|---|---|---|---|---|
| (a,b) | 0 | 25.5 | 32.5 | 22 |
| c | 25.5 | 0 | 28 | 39 |
| d | 32.5 | 28 | 0 | 43 |
| e | 22 | 39 | 43 | 0 |
now the smallest distance is \(D_2 ((a,b),e)=22\)
Update again
New distances
\[ \begin{aligned} D_3(((a,b),e),c)= & \frac{D_2((a,b),c) + D_2(e,c)}{2}=\frac{25.5 + 39}{2}=32.25\\ D_3(((a,b),e),d)= & \frac{D_2((a,b),d) + D_2(e,d)}{2}=\frac{32.5 + 43}{2}=37.75 \end{aligned} \]
| ((a,b),e) | c | d | |
|---|---|---|---|
| ((a,b),e) | 0 | 32.25 | 37.75 |
| c | 32.25 | 0 | 28 |
| d | 37.75 | 28 | 0 |
and again
new distance matrix
\[ \begin{aligned} D_4((c,d),((a,b),e)) & = \frac{D_3(c,((a,b),e)) + D_3(d,((a,b),e))}{2}\\ & = \frac{32.25+37.75}{2}\\ & =35 \end{aligned} \]
| ((a,b),e) | (c,d) | |
|---|---|---|
| ((a,b),e) | 0 | 35 |
| (c,d) | 35 | 0 |
finally
This is hierarchical clustering
This is called average linking, or Weighted Pair Group Method with Arithmetic Mean
One problem: we mix groups of different size
Node ((a,b),e) has three sequences, and (c,d) has two
“bigger nodes” should have more weight
Alternative: UPGMA
Unweighted pair group method with arithmetic mean
The distance clusters \(\mathcal{A}\) and \(\mathcal{B}\), each of size \({|\mathcal{A}|}\) and \({|\mathcal{B}|}\), is the average of all distances \(d(x,y)\) between pairs of objects in \(\mathcal{A}\) and in \(\mathcal{B}\)
\[d_{(\mathcal{A} \cup \mathcal{B}),X} = \frac{|\mathcal{A}| \cdot d_{\mathcal{A},X} + |\mathcal{B}| \cdot d_{\mathcal{B},X}}{|\mathcal{A}| + |\mathcal{B}|}\]
Example
\[ \begin{aligned} D_2((a,b),c)& =\frac{D_1(a,c) \times 1 + D_1(b,c) \times 1)}{1+1}=\frac{21+30}{2}=25.5\\ D_2((a,b),d)& =\frac{D_1(a,d) + D_1(b,d)}{2}=\frac{31+34}{2}=32.5\\ D_2((a,b),e)& =\frac{D_1(a,e) + D_1(b,e)}{2}=\frac{23+21}{2}=22 \end{aligned} \] This is the same as before
Then
\(D_3(((a,b),e),c)=\frac{D_2((a,b),c) \times 2 + D_2(e,c) \times 1}{2+1}=\frac{25.5 \times 2 + 39 \times 1}{3}=30\) \(D_3(((a,b),e),d)=\frac{D_2((a,b),d) \times 2 + D_2(e,d) \times 1}{2+1}=\frac{32.5 \times 2 + 43 \times 1}{3}=36\)
| ((a,b),e) | c | d | |
|---|---|---|---|
| ((a,b),e) | 0 | 30 | 36 |
| c | 30 | 0 | 28 |
| d | 36 | 28 | 0 |
Comparison


Distance to internal nodes
One problem with these approaches is that all pairs are joined at the same distance from the common ancestor
But the mutation rate may be different for different branches
Minimization problem
If we know the topology, we can find the branch lengths
We minimize the squared difference between observed distance and tree distance
\[\min_{d_{ij}} \sum_i\sum_j(D_{ij}-d_{ij})^2\]
But we still need to find the tree topology
Neighbor joining
This is one of the most recommended methods
Instead of joining the nearest nodes in the distance matrix,
we look into a new matrix \(Q\)
\[Q_{ij} = (n-2) D_{ij} -\sum_k D_{ik} -\sum_k D_{kj}\]
This “normalized” value can be negative
Neighbor joining

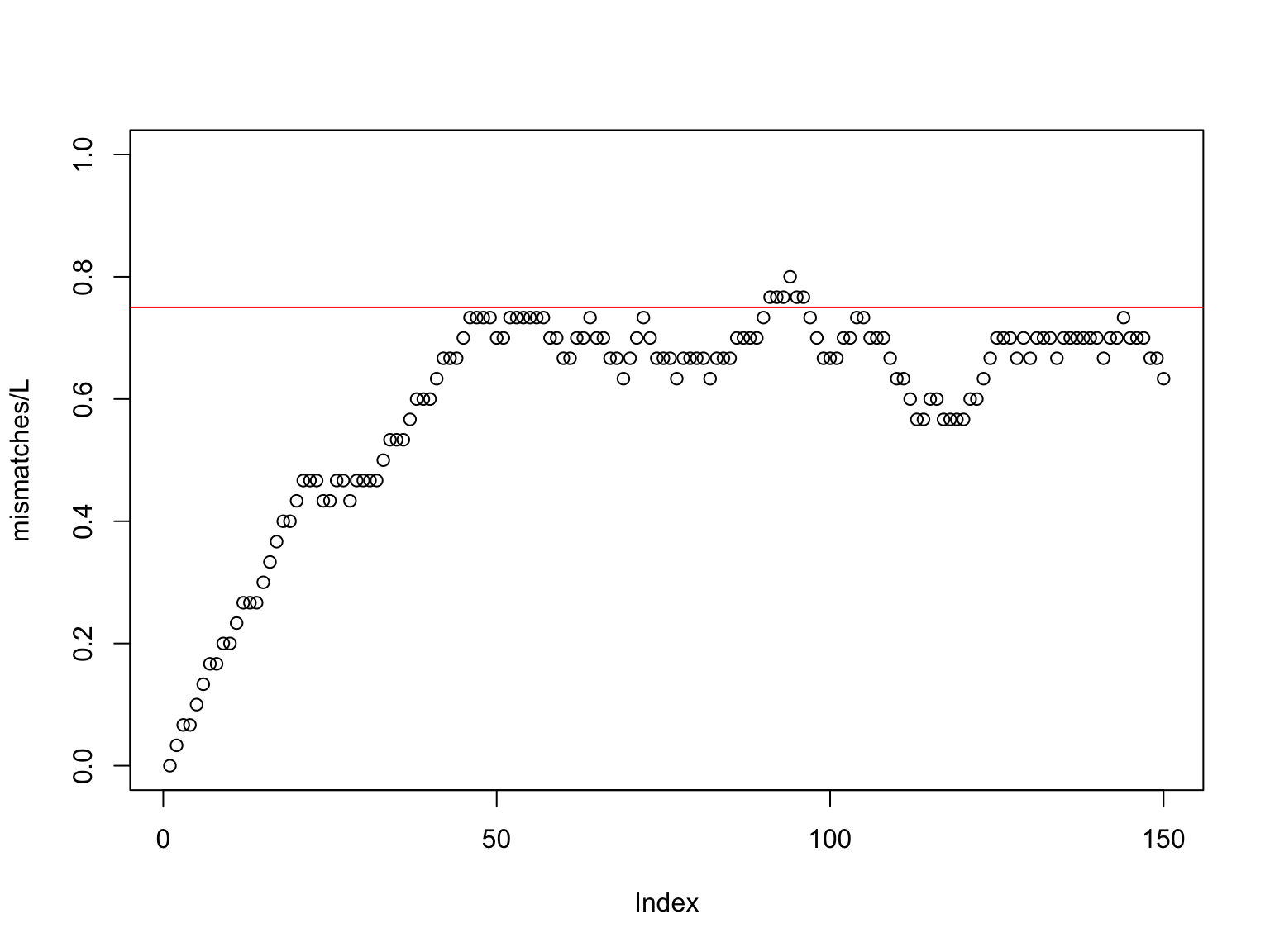
Distance is not time
Mutation rate is not proportional to time
Multiple substitutions cannot be observed
TATCGACTTCGGCAT
TATCGACGTCGGCAT
TATCGACTTCGGCAT
TATCGACTACGGCAT
TATCGACTTCGGCATSo we underestimate the divergence time
Max DNA mutation ≈ 75%
Substitution model
There are different models to find time given distance
The simplest one is Jukes-Cantor (1969)
\[Time = -\frac{3}{4}\ln\left(1-\frac{4}{3}Mismatch\right)\]
Here \(Mismatch\) is the percentage of sites with different nucleotides.
In summary
Making time machines is hard, and we only get an approximate answer