May 4th, 2018
DNA sequencing & assembly
How to sequence your own DNA
Sanger method
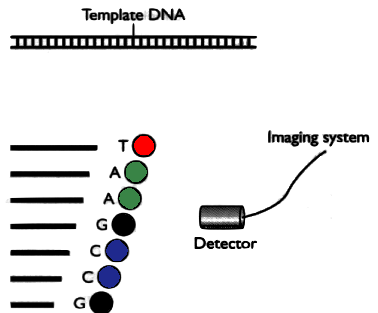
- Several DNA copies are cut using 4 restriction enzymes
- They cut specifically on each type of nucleotide
- They attach a fluorescent marker to the end
The detector sees different colors
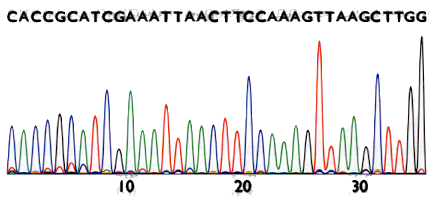
Fragments are separated by electrophoresis
Fragment length tells us the position of the nucleotide
The result is called chromatogram
From it we get the letters of the sequence
Shotgun DNA Sequencing
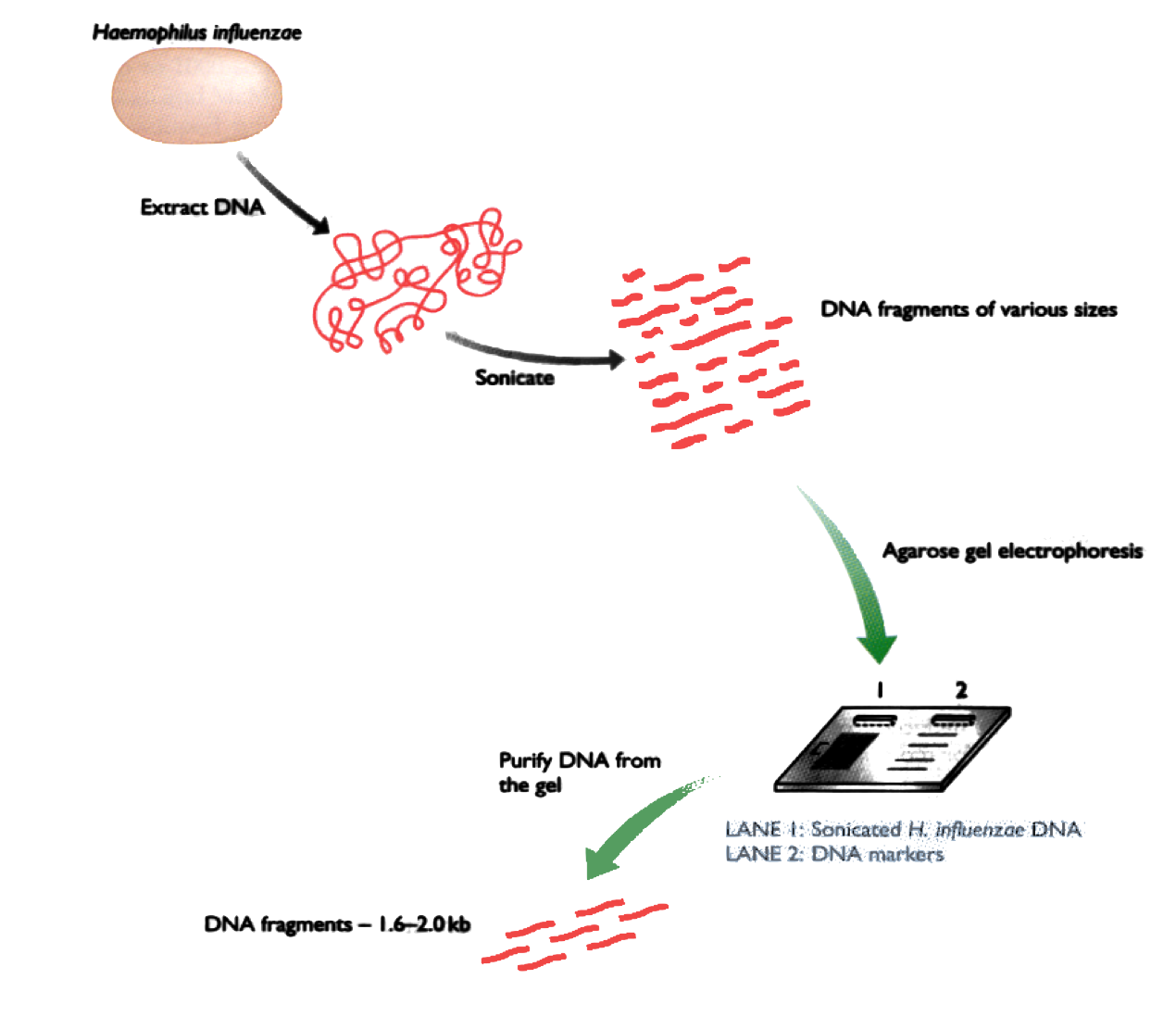
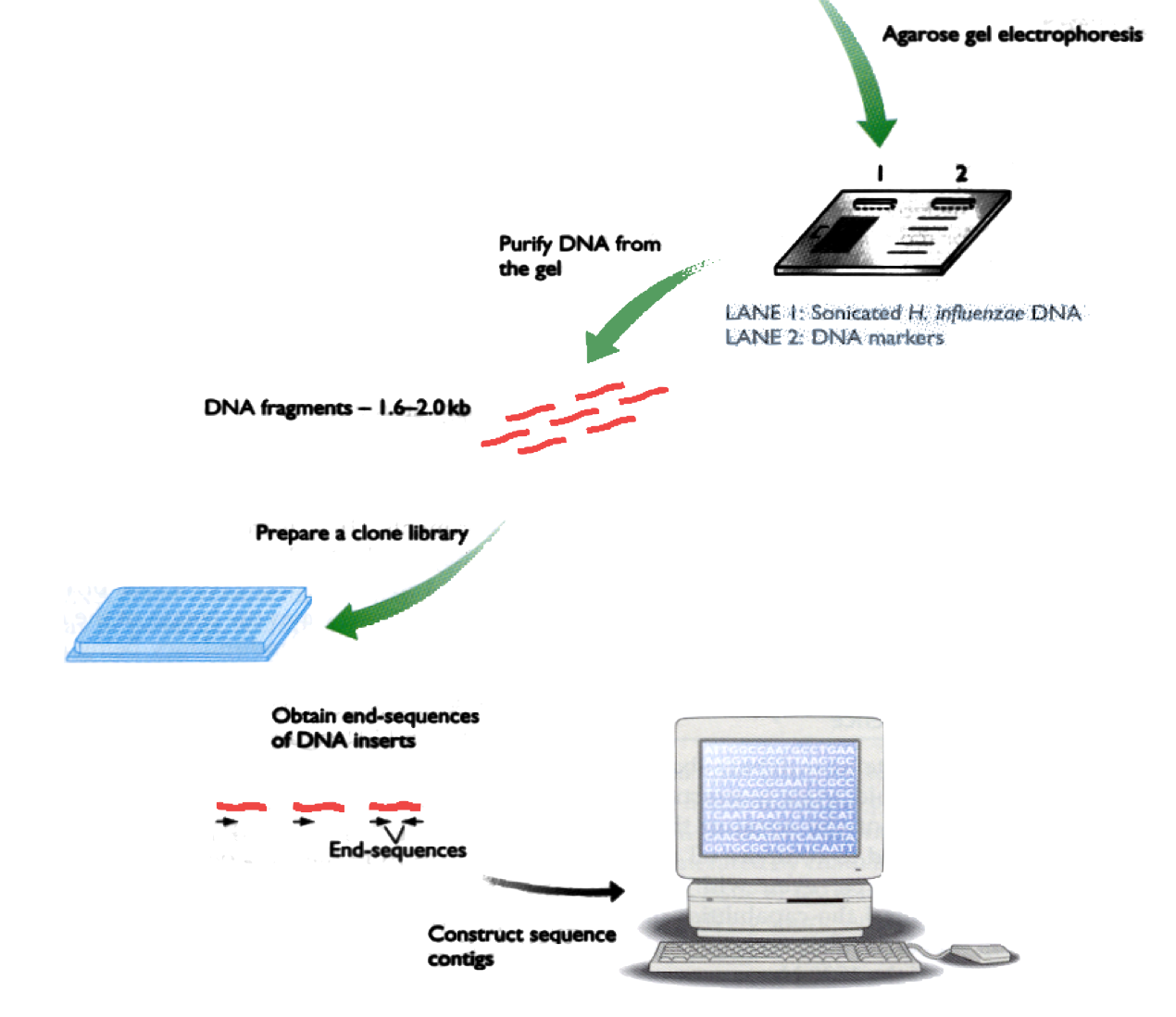
Shotgun DNA Sequencing (2)
Technology changes fast

In 2001, the cost of sequencing the first human genome was USD 108
Today you can have your own genome for 1000 USD
The problem is no longer how to do the experiment
Instead is how do we make sense of the results
DNA sequencing is cheap

A DNA sequencer in every desktop
First computers where big and expensive
Only in a few universities, used by experts
Then there was one on every office… and home
Today everybody has one… in the pocket
A PlayStation has more power than the biggest computer of 1998
Can the same happen with DNA sequencing?
The next iPhone

Today you can buy a DNA sequencer of the size of an iPhone
… at the price of an iPhone
Next step: people will make apps for DNA sequencer
Computing power grows exponentially
Moore’s Law

Big Picture
- Sequencing cost is going down
- Moore’s law: computing is cheap
There is a phase transition: we changed from “solid” to “liquid”

- Rules are changing very fast
For example, patents are obsolete
Producing data is cheap
There is already a lot of public data
- Tara Oceans published 7.8 Terabytes of metagenomic data
- equivalent to 1.660 DVDs
- Anybody can discover new knowledge there
- You can do it!
- And also many other people
This is a race
It does not depend on hands and wallets
It depends on brains and guts
All Science is Data Science
But Data Science is not about Data
Science is about obtaining
- Information
- Knowledge
- Wisdom
Genome Assembly
The sequencer produces small pieces
Current technology allows us to read DNA in runs of ~100-600 letters. Imagine a book of 1000 pages:
- several copies of the book are cut randomly in one million pieces
- different pieces may overlap
- half of the pieces are lost
- the remaining half is splashed with ink in the middle
The problem is to reconstruct the original book
In pictures

The protagonists of the story
- Genome: Only one, length
G, let’s say \(10^7\)
G <- 1E7
- reads:
Nof them, let’s say \(10^4\), each of lengthL
N <- 1E4 L <- 300
In Pictures
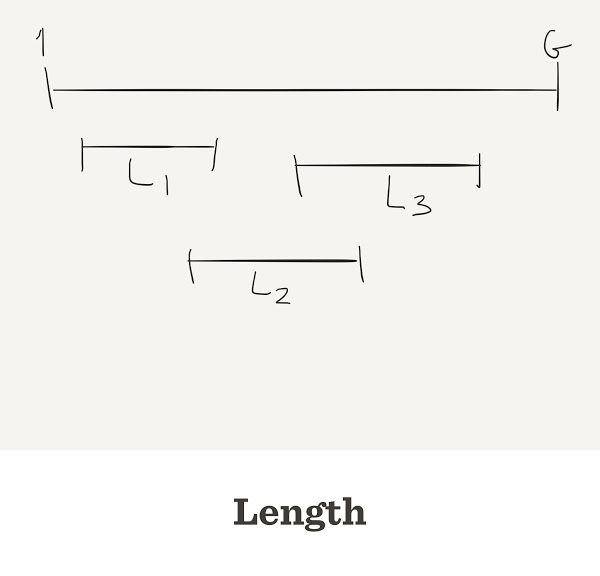
Reads are randomly taken from the genome
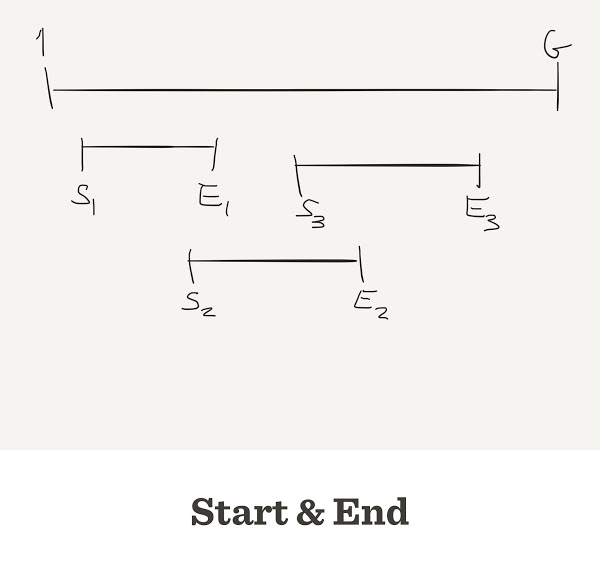
- Each read has a
startandendposition on the genome Lmust be equal toend-start- We take
startrandomly, then we calculateend
start <- sample.int(G, size=N, replace=TRUE) end <- start + L
Each read has start and end
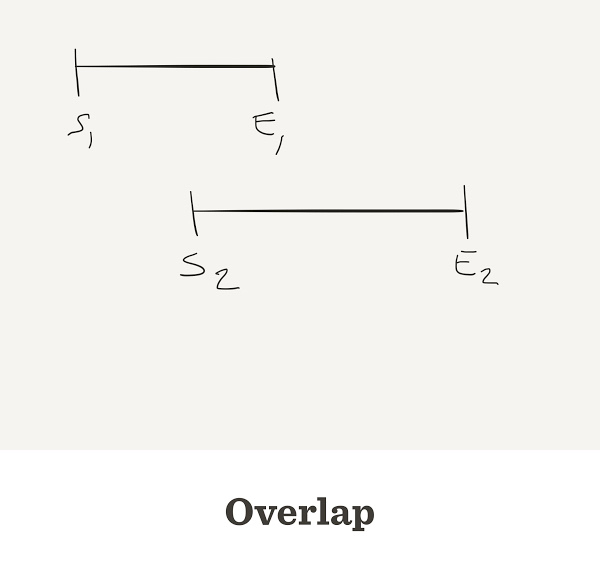
Sometimes reads do overlap
Sometimes they do not overlap
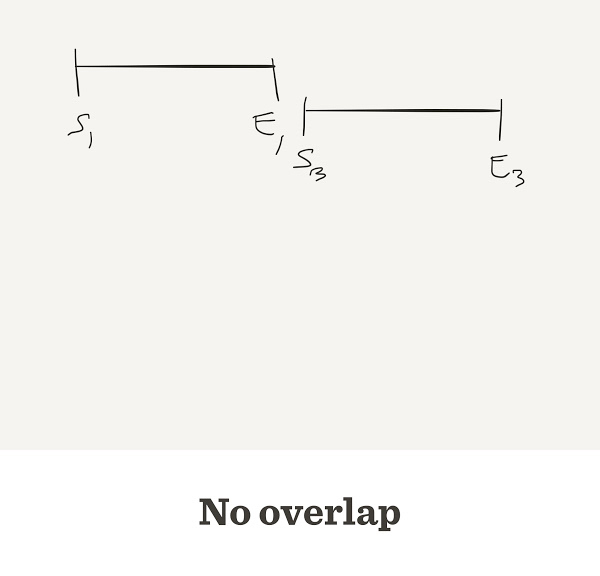
How do we know when two reads overlap?
How do we know when two reads overlap?
This is an event. That is, a function that returns TRUE or FALSE
- What are the inputs
- When must it return TRUE?
What is the overlap size?
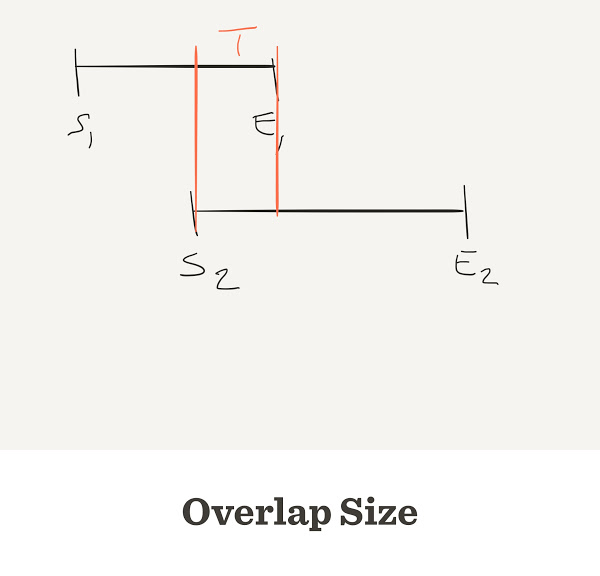
Negative gap size = overlap
If two reads do overlap, the overlap will be negative
Thus overlap_size is -gap_size
If we do not know their position, how can we detect if two reads do overlap?
Distribution of gap length

We only see overlaps over a threshold T
The assembler needs to compare all reads to all reads and see if they overlap.
- Can we detect overlap of 1 nucleotide?
- What is the best threshold
T?
If T is too small we get wrong results
If we decide that two reads overlap only when they share 1 letter, we will put together 25% of them every time
The best T has to be big enough to guarantee that the reads do not overlap by chance
If the reads do not match by chance, then there is a biological reason
Bigger values of T reduce the probability of “overlap by chance”
If two reads overlap, they are in the same Contig
A group of contiguous sequences is called Contig
The goal of the assembly process is to find one contig.
The sequence of this contig will be the genome
Most of times we do get several contigs