October 16, 2018
Your homework answers
about NGS
Congratulations
- Everybody delivered
- Everybody said something interesting/original
- Everybody made different mistakes
- Together, you can have the “big picture”
I made a mix of all your answers so everybody can see the good and bad
What is next generation sequence?
Buşra
Next-generation sequencing has a potential to dramatically accelerate biological research, by enabling the comprehensive analysis of genomes, transcriptomes and DNA-protein interactions to become inexpensive, routine and widespread
The next-generation sequencing technologies offer novel and rapid ways for genome-wide characterization and profiling of mRNAs, small RNAs, transcription factor binding regions, structure of chromatin and DNA methylation patterns, ancient DNA, microbiology and metagenomics
This review discusses next generation sequencing technologies, recent advances in next generation sequencing systems and applications for next generation technologies
Amplicon Generation
Buşra
Amplicon sequencing is a highly targeted approach that enables researchers to analyze genetic variation in specific genomic regions
The ultra-deep sequencing of PCR products (amplicons) allows efficient variant identification and characterization
This method uses oligonucleotide probes designed to target and capture regions of interest, followed by next-generation sequencing (NGS).
Amplicon sequencing is useful for the discovery of rare somatic mutations in complex samples (such as tumors mixed with germline DNA)
Another common application is sequencing the bacterial 16S rRNA gene across multiple species, a widely used method for phylogeny and taxonomy studies, particularly in diverse metagenomics samples
Advantages
Buşra
Enables researchers to efficiently discover, validate, and screen genetic variants using a highly targeted approach
Supports multiplexing of hundreds to thousands of amplicons per reaction to achieve high coverage
Delivers highly targeted re-sequencing even in difficult-to-sequence areas, such as GC-rich regions
Allows flexibility for a wide range of experimental designs
Reduces sequencing costs and turnaround time compared to broader approaches such as whole-genome sequencing
In Turkey
Buşra
Gen Era biotechnology i. company make amplicon generation in Istanbul
The price the all of genome 3000 euro
Roche 454
Ayşenur
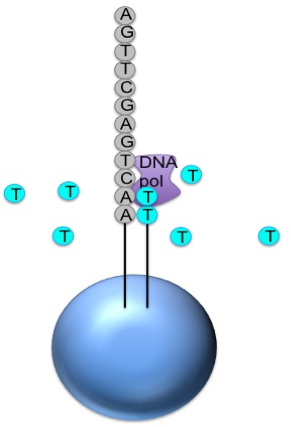 Generic adaptors are added to the ends and these are annealed to beads, one DNA fragment per bead
Generic adaptors are added to the ends and these are annealed to beads, one DNA fragment per bead
The fragments are then amplified by PCR using adaptor-specifc primers
Each bead is then placed in a single well of a slide
So each well will contain a single bead, covered in many PCR copies of a single sequence
The slide is flooded with one of the four NTP species
Roche 454
Ayşenur
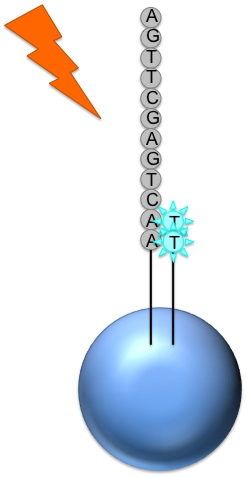 Where this nucleotide is next in the sequence, it is added to the sequence read
Where this nucleotide is next in the sequence, it is added to the sequence read
If that single base repeats, then more will be added
So if we flood with Guanine bases, and the next in a sequence is G, one G will be added, however if the next part of the sequence is GGGG, then four Gs will be added.
The addition of each nucleotide releases a light signal
These locations of signals are detected and used to determine which beads the nucleotides are added to
Roche 454
Ayşenur
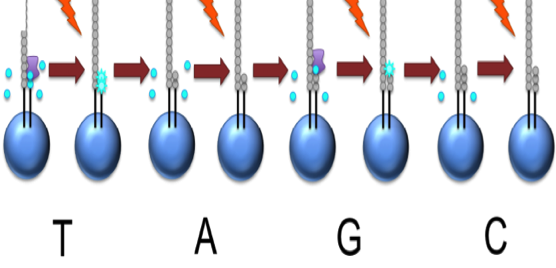 This NTP mix is washed away
This NTP mix is washed away
The next NTP mix is now added and the process repeated, cycling through the four NTPs.
This kind of sequencing generates graphs for each sequence read, showing the signal density for each nucleotide wash
The sequence can then be determined computationally from the signal density in each wash.
Roche 454
Ayşenur
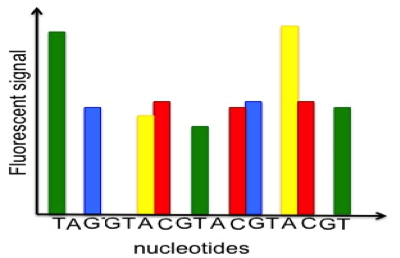 All of the sequence reads we get from 454 will be different lengths, because different numbers of bases will be added with each cycle.
All of the sequence reads we get from 454 will be different lengths, because different numbers of bases will be added with each cycle.
Roche 454
Ayşenur
- Advantage
- Read length, fast
- Disadvantage
- Error rate with polybase; high cost, low throughput
Roche 454
Eda
sequencing by synthesis
can sequence 0.7 gigabase per run
run time 23 hours
read lenght 700-800 bases
Roche 454
Faruk
Roche 454 pyrosequencing by synthesis (SBS) was the first commercially successful second generation sequencing system developed by 454 Life Sciences in 2005 and acquired by Roche in 2007 http://www.my454.com
This technology uses sequencing chemistry, whereby visible light is detected and measured after it is produced by an ATP sulfurylase, luciferase, DNA polymerase enzymatic system in proportion to the amount of pyrophosphate that is released during repeated nucleotide incorporation into the newly synthesized DNA chain
Roche 454
Faruk
The system was miniaturized and massively parallelized using PicoTiterPlates to produce more than 200,000 reads at 100 to 150 bp per read with an output of 20 Mb per run in 2005
The upgraded 454 GS FLX Titanium system released by Roche in 2008 improved the average read length to 700 bp with an accuracy of 99.997% and an output of 0.7 Gb of data per run within 24 h.
The GS Junior bench-top sequencer produced a read length of 700 bp with 70 Mb throughput and runtime of 10 to 18 h.
The major drawbacks of this technology are the high cost of reagents and high error rates in homopolymer repeats.
Roche 454
Faruk
The estimated cost per million bases is $10 by Roche 454 compared to $0.07 by Illumina HiSeq 2000.
A more serious challenge for those using this technology is the announcement by Roche that they will no longer supply or service the 454 sequencing machines or the pyrosequencing reagents and chemicals after 2016
Illumina Solexa
Eda
also sequencing by synthesis
- previous model Genome Analyzer can sequence 1Gb per run
- Solexa model can sequence 600 Gb per run
- HiSeq model 100 bases read length average 30Gb per run
- 11 day in regular mode
- 2 day in rapid run mode
and IMPROVING…
Illumina
Esra
- ∼$1000 MiSeq
- ∼$3000 HighSeq
- ∼ 1.5 days
Illumina Solexa
Faruk
Illumina http://www.illumina.com purchased the Solexa Genome Analyzer in 2006 and commercialized it in 2007
Today, it is the most successful sequencing system with a claimed over 70% dominance of the market, particularly with the HiSeq and MiSeq platforms
The Illumina sequencer is different from the Roche 454 sequencer in that it adopted the technology of sequencing by synthesis using removable fluorescently labeled chain-terminating nucleotides that are able to produce a larger output at lower reagent cost
Illumina Solexa
Faruk
The clonally enriched template DNA for sequencing is generated by PCR bridge amplification (also known as cluster generation) into miniaturized colonies called polonies
The output of sequencing data per run is higher (600 Gb), the read lengths are shorter (approximately 100 bp), the cost is cheaper, and the run times are much longer (3-10 days) than most other systems [54]
Illumina Solexa
Faruk
Illumina provides six industrial-level sequencing machines (NextSeq 500, HiSeq series 2500, 3000, and 4000, and HiSeq X series five and ten) with mid to high output (120–1500 Gb) as well as a compact laboratory sequencer called the MiSeq, which, although small in size, has an output of 0.3 to 15 Gb and fast turnover rates suitable for targeted sequencing for clinical and small laboratory applications
The MiSeq uses the same sequencing and polony technology such as the high-end machines, but it can provide sequencing results in 1 to 2 days at much reduced cost
Illumina’s new method of synthetic long reads using TruSeq technology apparently improves de novo assembly and resolves complex, highly repetitive transposable elements
ABI-SOLID
Eda
sequencing by ligation
- Solid 4 model can read 80-100 Gb per run (coverage 30Gb)
- average read length 50 bases
- fragment length 400-600-bp
ABI-SOLID
Faruk
Supported Oligonucleotide Ligation and Detection (SOLiD) is a next-generation sequencer instrument marketed by Life Technologies http://www.lifetechnologies.com and first released in 2008 by Applied Biosystems Instruments (ABI)
It is based on 2-nucleotide sequencing by ligation (SBL)
This procedure involves sequential annealing of probes to the template and their subsequent ligation
ABI-SOLID
Faruk
Sequencers on the market today, such as the 5500 W series, are suitable for small- and large-scale projects involving whole genomes, exomes, and transcriptomes
Previously, sample preparation and amplification was similar to that of Roche 454 sequencing
However, the upgrades to Wildfire chemistry have enabled greater throughput and simpler workflows by replacing beads with direct in situ amplification on FlowChips and paired-end sequencing
ABI-SOLID
Faruk
The SOLiD 5500 W series sequencing reactions still use fluorescently labeled octamer probes in repeated cycles of annealing and ligation that are interrogated and eventually deciphered in a complex subtractive process using Exact CallChemistry that has been well described by others
The advantage of this method is accuracy with each base interrogated twice
The major disadvantages are the short read lengths (50–75 bp), the very long run times of 7 to 14 days, and the need for state-of-the-art computational infrastructure and expert computing personnel for analysis of the raw data
DNA nanoball sequencing by BGI Retrovolocity
Faruk
Complete Genomics http://www.completegenomics.com developed DNA nanoball sequencing (DNBS) as a hybrid of sequencing by hybridization and ligation
Small fragments (440–500 bp) of genomic DNA or cDNA are amplified into DNA nanoballs by rolling-circle replication that requires the construction of complete circular templates before the generation of nanoballs
The DNA nanoballs are deposited onto an arrayed flow cell, with one nanoball per well sequenced at high density
Up to 10 bases of the template are read in 5′ and 3′ direction from each adapter
DNA nanoball sequencing by BGI Retrovolocity
Faruk
Since only short sequences, adjacent to adapters, are read, this sequencing format resembles a multiplexed form of mate-pair sequencing similar to using Exact Call Chemistry in SOLiD sequencing
Ligated sequencing probes are removed, and a new pool of probes is added, specific for different interrogated positions
The cycle of annealing, ligation, washing, and image recording is repeated for all 10 positions adjacent to one terminus of one adapter
This process is repeated for all seven remaining adapter termini
DNA nanoball sequencing by BGI Retrovolocity
Faruk
Although the developers have sequenced the whole human genome, the major disadvantage of DNBS is the short length of reads and the length of time for the sequencing projects
Claimed cost of the reagents for sequencing of the whole human genome is under $5000
The major advantage of this approach is the high density of arrays and therefore the high number of DNBs (~350 million) that can be sequenced
DNA nanoball sequencing by BGI Retrovolocity
Faruk
In 2015, the Chinese genomics service company BGI-Shenzhen acquired Complete Genomics and introduced the Retrovolocity system for large-scale, high quality whole-genome and whole-exome sequencing with 50x coverage per genome and with the sample to assembled genome produced in less than 8 days
Complete Genomics claims to have sequenced more than 20,000 whole human genomes over 5 years and published widely on the use of their NGS platform
They provide public access to a human repository of 69 genomes data and a cancer data set of two matched tumor and normal sample pairs at http://www.completegenomics.com/public-data/
Ion torrent
Faruk
Ion Torrent technology http://www.iontorrent.com was developed by the inventors of 454 sequencing, introducing two major changes.
Firstly, the nucleotide sequences are detected electronically by changes in the pH of the surrounding solution proportional to the number of incorporated nucleotides rather than by the generation of light and detection using optical components.
Ion torrent
Faruk
Secondly, the sequencing reaction is performed within a microchip that is amalgamated with flow cells and electronic sensors at the bottom of each cell
The incorporated nucleotide is converted to an electronic signal detected by the electronic sensors
Ion torrent
Faruk
The two sequencers in the market that use Ion Torrent technology are the high-throughput Proton sequencer with more than 165 million sensors and the Ion Personal Genome Machine (PGM), a bench-top sequencer with 11.1 million sensors
Ion torrent
Faruk
There are four sequencing chips to choose from
The Ion PI Chip is used with the Proton sequencer, and the Ion 314, 316, or 318 Chips are used with the Ion PGM.
The Ion 314 Chip provides the lowest reads at 0.5 million reads per chip, whereas the Ion 318 Chip provides the highest reads of up to 5.5 million reads per chip
Ion torrent
Faruk
The Proton sequencer provides a higher throughput (10–100 Gb vs. 20 Mb–1 Gb) and more reads per run (660 Mb vs. 11 Mb) than the PGM chips, but the read lengths (200–500 bp), run time (4–5 h), and accuracy (99%) are similar
Sample preparation for the generation of DNA libraries is similar to the one used for Roche 454 sequencing but can be simplified with the use of the Ion Chef system for automated template preparation and chip loading
Ion torrent
Faruk
The Ion Torrent chip is used with an ion-sensitive field-effect transistor sensor that has been engineered to detect individual protons produced during the sequencing reaction
The chip is placed within the flow cell and is sequentially flushed with individual unlabeled dNTPs in the presence of the DNA polymerase
Ion torrent
Faruk
Incorporation of nucleotide into the DNA chain releases H protons and changes the pH of the surrounding solution that is proportional to the number of incorporated nucleotides
The major disadvantages of the system are problems in reading homopolymer stretches and repeats
The major advantages seem to be the relatively longer read lengths, flexible workflow, reduced turnaround time, and a cheaper price than those provided by the other platforms.
PacBio
Atakan
- PacBio model RS II produces average read lengths over 10 kb, with an N50 of more than 20 kb and maximum read lengths over 60 kb
- Error rate of a continuous long read is relatively high (around 11%–15%)
- The error rate can be reduced by generating circular consensus sequences reads with sufficient sequencing passes.
Summary
Atakan
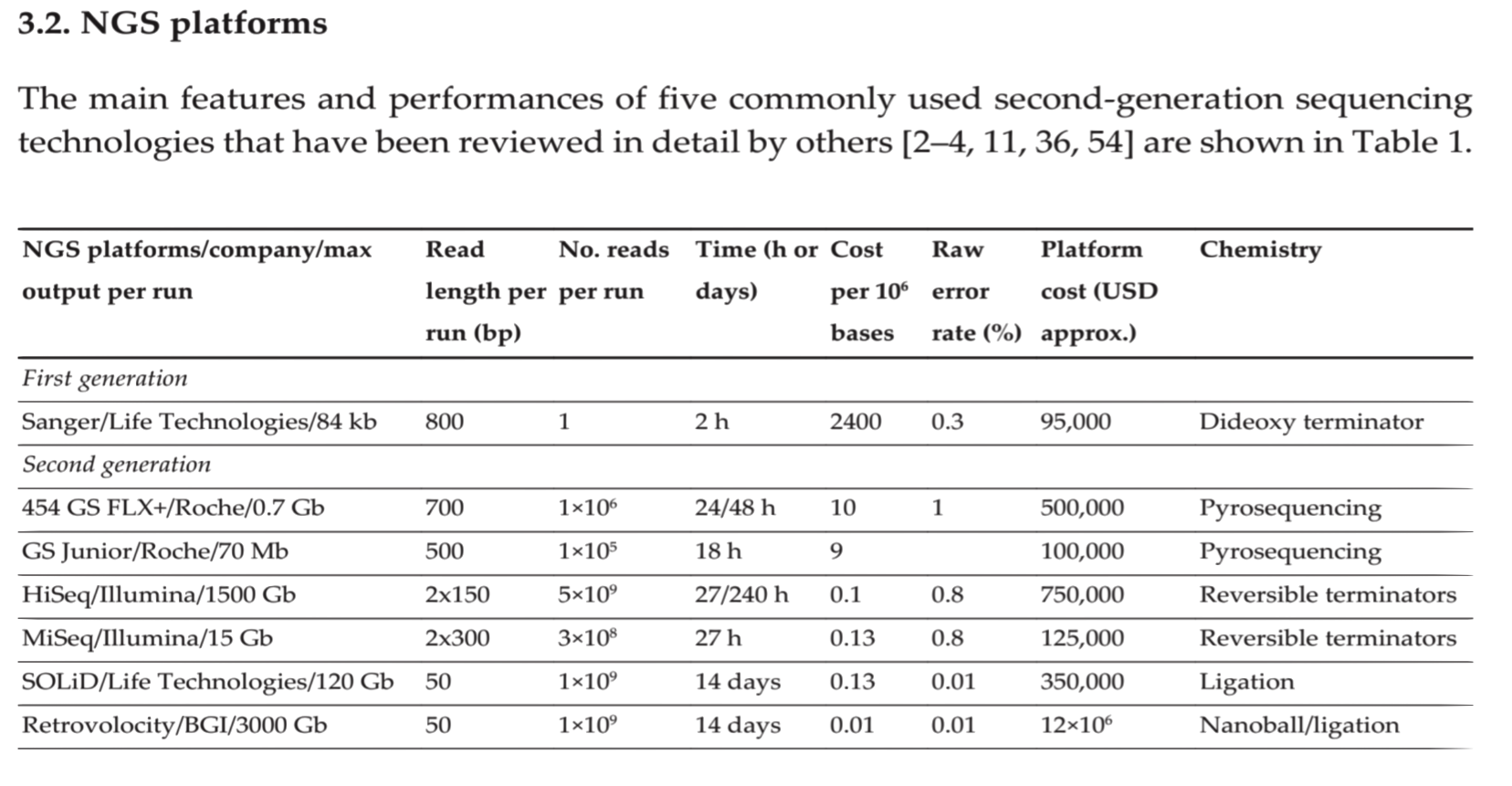
| Method | Generation | Read length (bp) Single pass | Error rate (%) | No. of reads per run | Time per run | Cost per million bases (USD) |
|---|---|---|---|---|---|---|
| Sanger ABI 3730x1 | 1st | 600-1000 | 0.001 | 96 | 0.5 - 3 h | 500 |
| Illumina HiSeq 2500 (Rapid Run) | 2nd | 2 x 250 | 0.1 | 1.2 x 109 (paired) | 1 - 6 days | 0.04 |
| PacBio RS II: P6-C4 | 3rd | 1.0–1.5 x 104 | 13 | 3.5–7.5 x 104 | 0.5 - 4 h | 0.4 - 0.8 |
| Oxford Nanopore MinION | 3rd | 2–5 x 103 | 38 | 1.1–4.7 x 104 | 50 h | 6.44 - 17.9 |
Summary
Esra
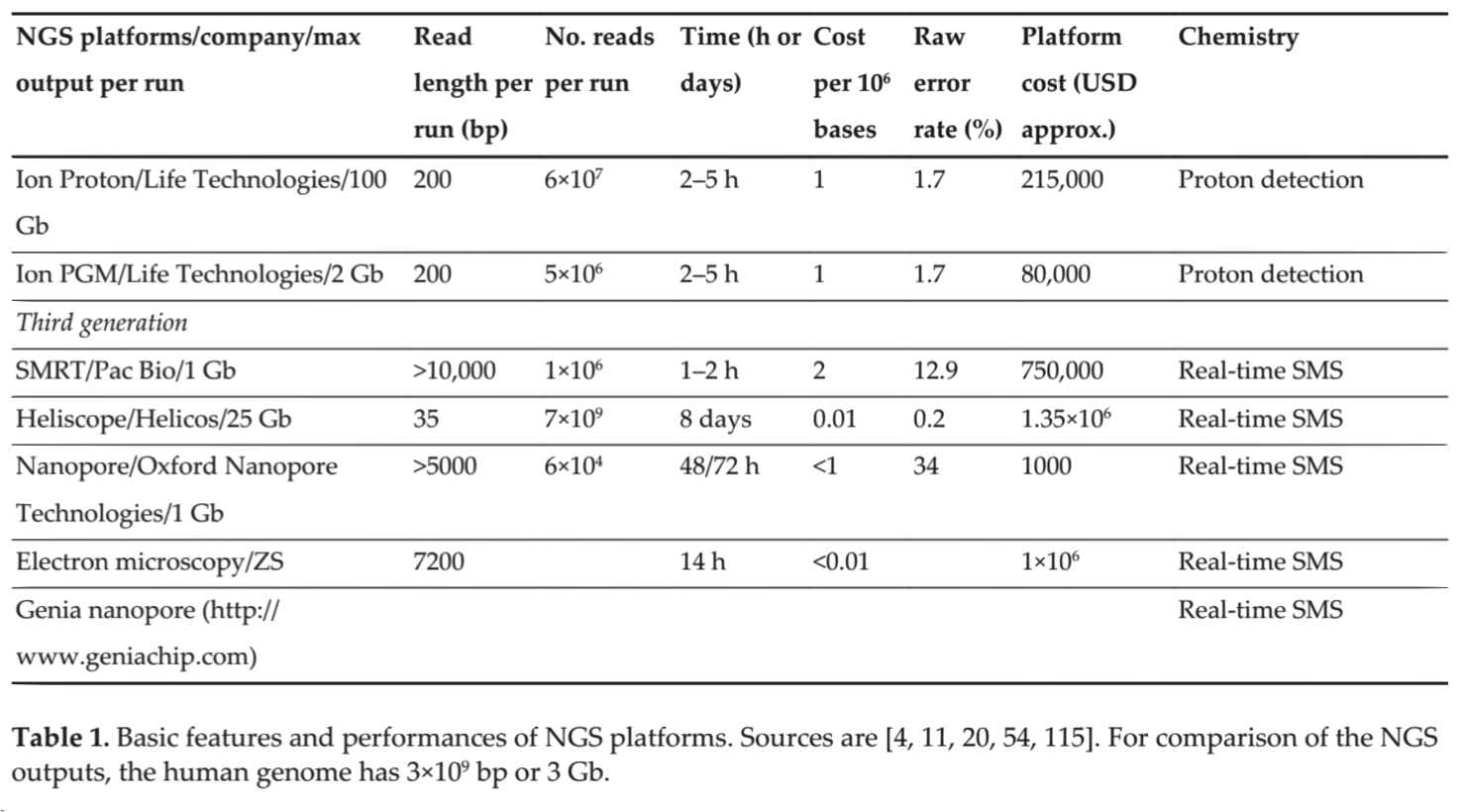
| Instrument | Primary Errors | Single-pass Error Rate (%) | Final Error Rate (%) |
|---|---|---|---|
| 3730xl (capillary) | substitution | 0.1-1 | 0.1-1 |
| 454 All models | Indel | 1 | 1 |
| Illumina All Models | substitution | ~0.1 | ~0.1 |
| Ion Torrent – all chips | Indel | ~1 | ~1 |
| SOLiD – 5500xl | A-T bias | ~5 | ≤0.1 |
| Oxford Nanopore | deletions | ≥4 | 4 |
| PacBio RS | Indel | ~13 | ≤1 |
Summary
Faruk
Summary
Faruk
References
- Faruk
- Jerzy K. Kulski Next-Generation Sequencing — An Overview of the History, Tools, and “Omic” Applications (2016) DOI: 10.5772/61964
- Esra
- Glenn et al. Molecular Ecology Resources, 2011 May, doi.org/10.1111/j.1755-0998.2011.03024.x
- ChIPBase, School of Life Science, Sun Yat-sen University, China.
- ODIN, detecting differential peaks in ChIP-Seq. DOI: 10.1093/bioinformatics/btu722.
References
- Esra
- Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data, https://doi.org/10.1371/journal.pcbi.1003326
- Probabilistic Inference on Multiple Normalized Signal Profiles from Next Generation Sequencing: Transcription Factor Binding Sites, https://doi.org/10.1109/TCBB.2015.2424421
- SignalSpider: probabilistic pattern discovery on multiple normalized ChIP-Seq signal profiles, https://doi.org/10.1093/bioinformatics/btu604
My comments
- Always include references to your sources. This is essential in science
- Not doing it is unethical
- Doing it gives you credibility
- Summarize. Don’t make me think
- What do the reader needs to know
- Put yourself in the reader’s place
- So what? What are the consequences?
- How can we use your data?
- Information is the part of data that allows us to make a decision
Exam question
- You want to sequence an organism. You know the approximate genome size
- What technology will you use? Why?
- How long will it take?
- How many contigs do you expect?
- What will be the average depth?
- What will be the N50?
- How much will it cost?
Mapping reads to a reference
Mapping reads to a reference
As we saw in the last class, mapping reads to a reference genome is a useful technique
- It can be use to assemble a closely related organism
- It can be use to detect polymorphisms
What is mapping?
Mapping is finding where each read maps on the genome
Here “maps” means “the genome sequence is very similar to the read”
The idea is that
- if the genome is fragmented,
- and the read is part of the genome
then we want to know where the fragment came from
How do you find the place of the read?
For now, let’s assume that the read is 100% identical to a part of the genome
The problem is identical to “finding a word in a text”
How does CTRL-F works?
Direct way
for(i in 1:length(genome)) {
n <- 0
for(j in 1:length(read)) {
if(genome[i]==read[j]){
n <- n+1
}
}
if(n==length(read)){
print(i)
}
}
How long does it take? (time)
How long does it take?
The time of a program is usually called its computational cost
It depends on the genome length \(G\) and the read length \(L\)
There are two loops. The inner part of the loop is done \(G\cdot L\) times
We say that the cost of this program is \(O(GL)\)
Cost is “on the order of” \(G\cdot L\)
For example, for a genome of 3 million bp and a read of 500 bp, the cost is on the order of 1.5E9 steps
That is 1.500.000.000 steps for one read
It is worse
We have to look for several reads. Millions. Let’s say \(N\)
Then we have to repeat everything \(N\) times
If we have 2 million reads, we multiply 1.5E9 by 2E6
That is 3E15 = 3.000.000.000.000.000 steps
Assuming that the computer can do 1000 million steps per second, this is \[3\cdot 10^{15}\text{ steps}\cdot \frac{1\text{ sec}}{10^{9}\text{ steps}}\cdot\frac{1\text{ day}}{86400\text{ sec}}\approx 34.7\text{ days}\]
How can we make it faster?
 Clearly, a direct search is a bad idea
Clearly, a direct search is a bad idea
All modern systems use a speed-up technique based in some kind of index
Indices are an auxiliary table that show where we can find each “word” in the “text”
For example as we see at the end of a book
“Think Complexity” by Allen B. Downey CC-BY-NC-SA http://thinkcomplex.com/thinkcomplexity.pdf
What is “word” and “text”?
Most of the algorithms used in bioinformatics were initially created for other purposes
Here we are using ideas that were initially developed for searching patterns (such as words) inside long texts, like in a book
In our context, the text will be the full genome, and the words are subsequences of DNA of a fixed length.
Usually, the word length is called k, and the words are called k-mers
To look for a read in the genome, we see which k-mers are in the read, and we look for them first
Example: index of 2-mers
GATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTAT
| key | position | key | position |
|---|---|---|---|
| AA | 24 | GA | 1, 36 |
| AC | 5, 16, 25, 28, 32 | GC | 38, 47 |
| AG | 7, 37 | GG | 8, 34, 35, 53 |
| AT | 2, 13, 21, 45, 49, 56 | GT | 9, 54 |
| CA | 4, 6, 15, 27, 31, 44, 48 | TA | 12, 20, 23, 55 |
| CC | 17, 18, 26, 43 | TC | 3, 10, 14, 30, 40, 42 |
| CG | 33 | TG | 46, 52 |
| CT | 11, 19, 29, 39, 41 | TT | 22, 50, 51 |
When data is sorted, we can search much faster
Searching in the index is easy, because it is sorted
When data is sorted, we can search quickly by dividing the list in half every step
This is called Binary Search. Google it
Using this strategy of dividing the index in half on every step, we need \[\log_2(\text{Number of words in the index})\cdot (\text{word length})\] steps to find any word in the index
Computational cost of search in the index
It is easy to see that there are \(4^k\) different k-mers
Some may appear several times on the genome, some may not appear
On average, each k-mer appears \(G/4^k\) times on the genome
In any case, the number of words in the index is \(4^k\)
Using the strategy of binary search, we can find each word in the index
\[\log_2(4^k)\cdot k=\log_2(2^{2k})\cdot k=2k^2\]
Searching using the index
After we find the k-mer in the index, we have to see it the rest of the read is on the genome
This time we do not need to see all the genome
we have to look only \(G/4^k\) positions on the genome
As before, testing to see if the read is on any position takes \(L\) steps
The total cost of a single read is then \[2k^2\cdot\frac{G}{4^k}\cdot L\]
Example with k=4
For the same genome of 3 million bp, using k=4, the index will have 256 entries, each of length \[\frac{3\times 10^{6}}{4^4}\approx 1.17\times 10^{4}\] searching one read of 500bp will take \[2\cdot 4^2\cdot 1.17\times 10^{4} \cdot 500=1.88\times 10^{8}\] steps, that is, 0.19 seconds per read. For 2 million reads, this takes \[2\times 10^{6}\text{ reads}\cdot 0.19\frac{\text{sec}}{\text{read}}\cdot\frac{1\text{ day}}{86400\text{ sec}}\approx 4.34\text{ days}\]
Example with k=6
For the same genome of 3 million bp, using k=6, the index will have 4096 entries, each of length \[\frac{3\times 10^{6}}{4^6}\approx 732.422\] searching one read of 500bp will take \[2\cdot 6^2\cdot 732.422 \cdot 500=2.637\times 10^{7}\] steps, that is, 0.026 seconds per read. For 2 million reads, this takes \[2\times 10^{6}\text{ reads}\cdot 0.026\frac{\text{sec}}{\text{read}}\cdot\frac{1\text{ hour}}{3600\text{ sec}}\approx 14.648\text{ hours}\]
Example with k=8
For the same genome of 3 million bp, using k=8, the index will have \(6.554\times 10^{4}\) entries, each of length \[\frac{3\times 10^{6}}{4^8}\approx 45.776\] searching one read of 500bp will take \[2\cdot 8^2\cdot 45.776 \cdot 500=2.93\times 10^{6}\] steps, that is, 0.003 seconds per read. For 2 million reads, this takes \[2\times 10^{6}\text{ reads}\cdot 0.003\frac{\text{sec}}{\text{read}}\cdot\frac{1\text{ hour}}{3600\text{ sec}}\approx 1.628\text{ hours}\]
Nothing is free in life
As we can see, using an index is much faster. But we need to make and store the index
Can you estimate how many steps do we need to make the index?
Can you estimate how much memory is needed?
Obviously, the answer depends on the value of \(k\)