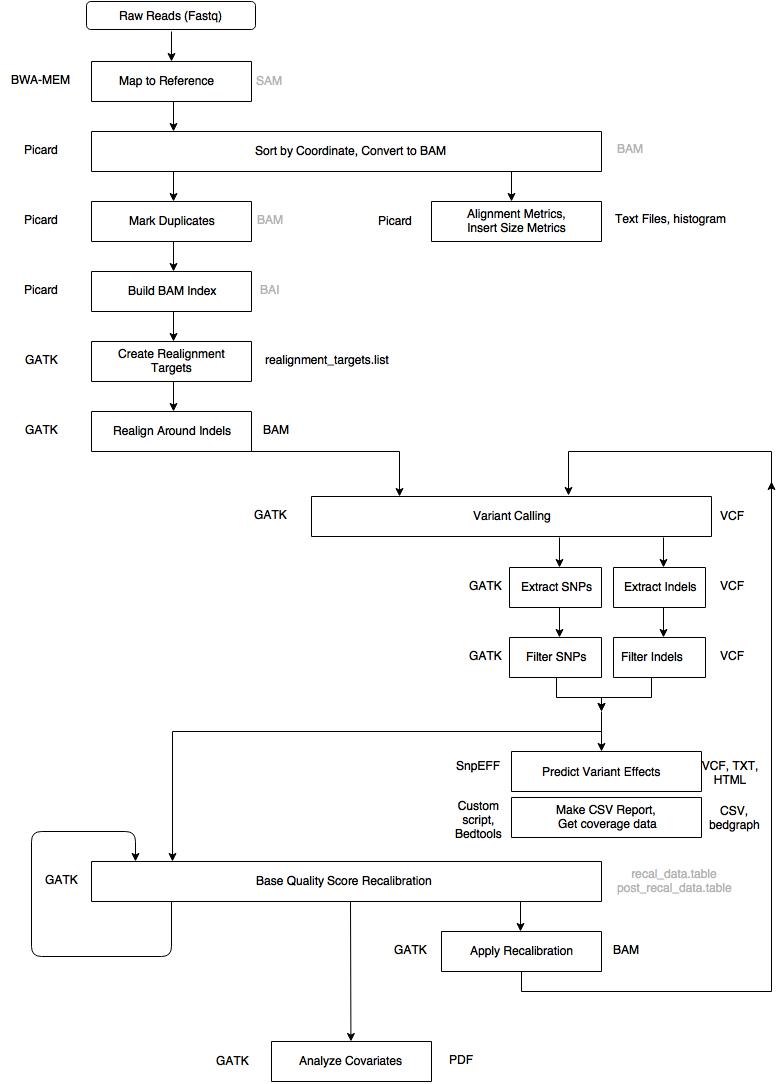
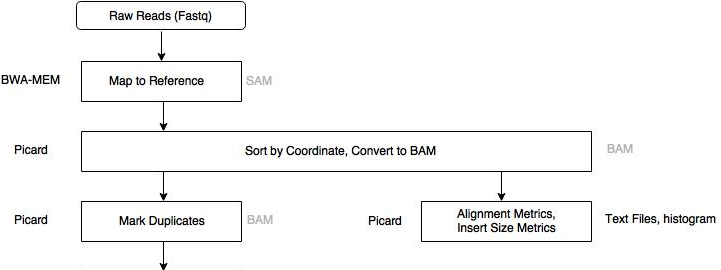
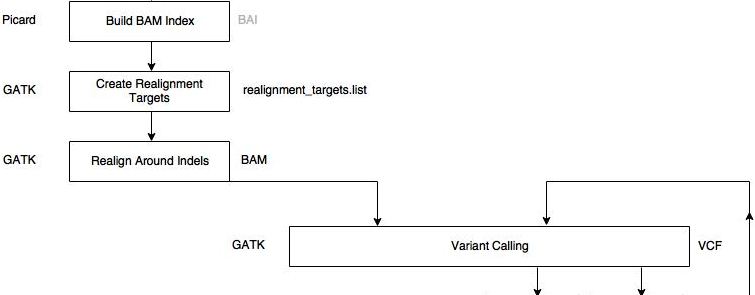
An application of bioinformatics in clinical context.
Determination of polymorphisms in genes associated with known diseases
A specific gene is amplified using PCR. The product is sequenced using NGS.
Bioinformatic analysis starts with the fastq files, and the human genome as reference