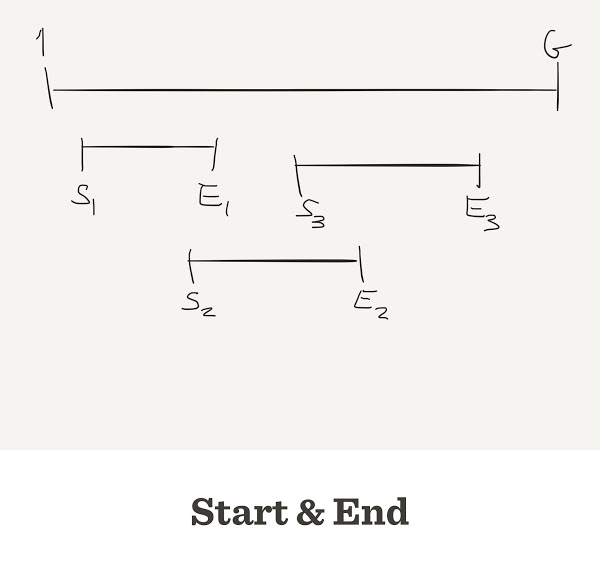
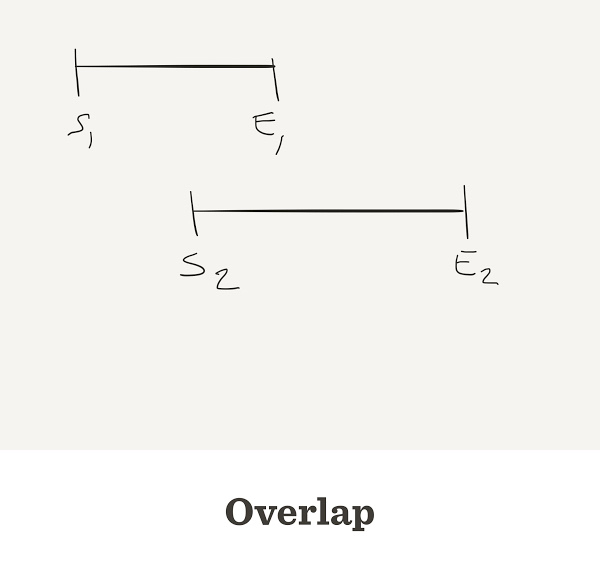
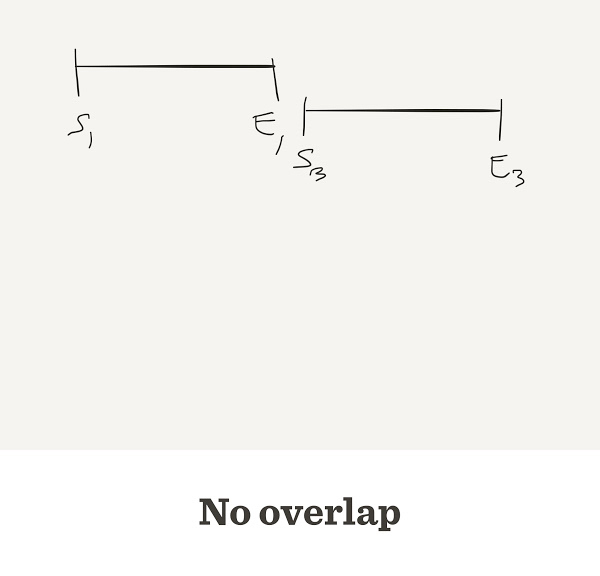
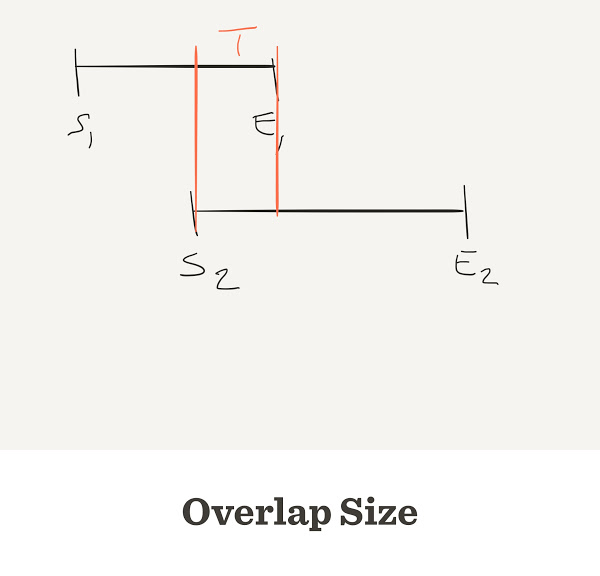
The number of contigs depends on
- Genome length:
G - Length of reads:
L - Number of reads:
N
In general L can be different for each read. In this simulation we will assume all reads have the same length
Exercise: Find the real values of L in our reads