Türkçe bilmiyorum 😟
I am
- Mathematical Engineer, U. of Chile
- PhD Informatics, U Rennes 1, France
- PhD Mathematical Modeling, U. of Chile
- not a Biologist
- but an Applied Mathematician who can speak “biologist language”
April 15, 2015
Türkçe bilmiyorum 😟
I am
Institute strongly connected to the Department of Mathematical Engineering. On 2013 it had:
42 Researchers
10 Postdoctoral Fellows
11 Laboratories
29 Phd students
85 ISI Publications
1087 Citations in ISI Journals
3 Patents
It was the first of 9 International Joint Unit of CNRS (France’s TUBITAK) on mathematics.

world

chile
Near 17 million people
Universities ranks similar to Turkish ones
Spanish colony 500 years ago (so language is Spanish)
Independent Republic 200 years ago
First Latin American country to recognize Turkish republic
OECD member, same as Turkey
Everyday life very similar to Turkey

exports
1st world producer of copper
2nd world producer of salmon
Fruits: peaches, grapes, apples, avocado
Wine: exported worldwide
Biotechnology can improve all these industries
Official data for 2014. Banco Central de Chile
Bioleaching is much better that melting copper
The goal is to understand and improve the involved bacteria so this technology can be used extensively
Enables building new mines
It is like discovering petrol reserves for the country
We had a research contract with the main mining company
State owned, big enough to pay for long term research
Few papers, many patents
In Biology every rule has an exception
A biological “law” is verified ~80% of time
Theory is in constant change
And yet it moves
Focus on the pieces that form these pieces. Things that can not be observed on the microscope
The tree of life has two main branches
This is the whole picture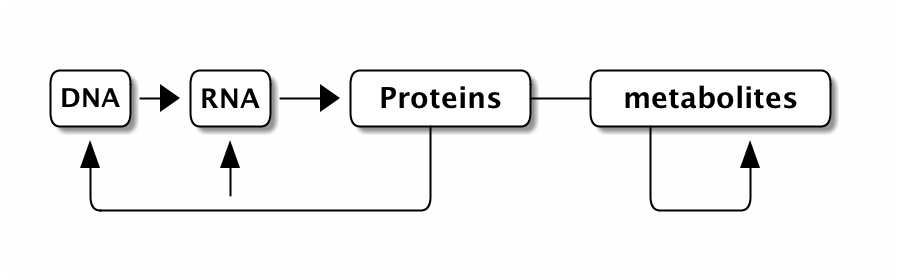
In some cases we focus only on the relation between DNA and proteins
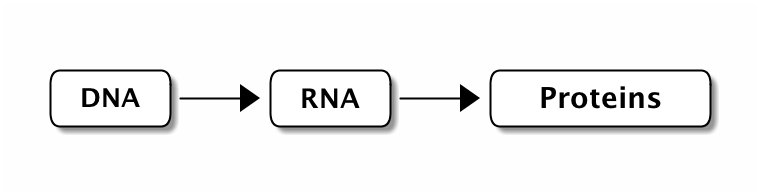
We can abstract the chemical nature of these molecules and look them as sequences of symbols
When the cell needs a protein
Each object involves a mathematical problem
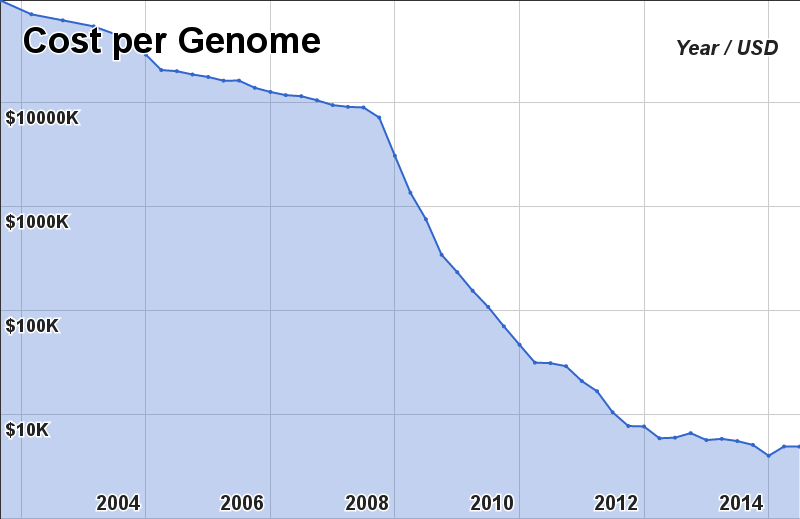
Current technology allows us to read DNA in runs of ~100-600 letters. Imagine a book of 1000 pages:
The problem is to reconstruct the original book
The classical approach is to see each “piece” as a vertex of a graph. There is an edge when the two “pieces” overlap.
Lander & Waterman (1988) proved that the expected number of connected components is
\[E( C) = N e^{-\frac{NL}{G}}\] where N is the number of “pieces”, L the average length of the “pieces”, and G is the length of the chromosome.
But experimental results did not match the theory. There was a wrong hypothesis.
It was assumed that sequence overlap corresponded to physical overlap.
But some genes have multiples copies in the genome. A kind of backup.
The question is: How to traverse the graph and reconstruct the original sequence?
Once the complete genome has been assembled we need to find the “words” in the text. There are no “spaces”.
The usual approach is to see genes as a realization of a Markov Chain, the intergenic region as another chain, and the transition between both controlled by a hidden Markov chain.
This is the Hidden Markov Model. In practice the problem is how to find the good parameters. Good in Prokaryotes, not so good in Eukaryotes.
If we have found all “words”, what is their meaning?
It is observed that most genes are homolog to genes on other species. This homology is determined by an edit distance. We can “transform” a gene into another by
Each edition has a cost. The distance is the minimal cost (Method of Smith & Waterman).
What is a “reasonable distance”?
To evaluate significance we need a “null hypothesis”.
Karlin & Altschul described a model for the expected number of sequences within a given distance using substitution and arbitrary scores.
The general problem including insertions and deletion has not been formally solved, although there are some “rule of thumb” approaches.
Now we are in condition to evaluate (partially) the state of the cell by measuring the concentration of RNA.
The expression of a gene is the concentration of the RNA transcribed from the gene.
There are several techniques to do that. Some are based on the chemistry of nucleic acids:
Gene expression experiments result in a matrix.
Each row is a gene, each column an experiment.
The problem is to find structures in this matrix.
Classical case: clustering of genes by linear correlation
But correlation may be non-linear: entropy based mutual information
\[\int_{Y}\int_{X} p(x,y) \log \frac{p(x,y)}{p(x)p(y)} dx\,dy\]
Analysis of gene expression shows that not all genes are expressed all times. Some genes are regulators. They enable or disable the expression of other genes.
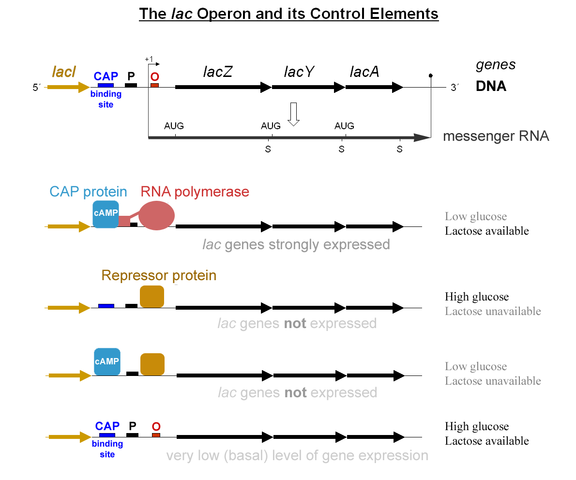
"Lac operon-2010-21-01" by Lac_operon.png: G3proderivative work: Tereseik (talk) - Lac_operon.png. Licensed under CC BY 2.0 via Wikimedia Commons - http://commons.wikimedia.org/wiki/File:Lac_operon-2010-21-01.png#/media/File:Lac_operon-2010-21-01.png
Determining true binding sites is hard. Current methods produce too many false positives.
Im my research I built a putative regulatory network for the well studied bacteria E.coli. We expected ~4K regulations. We got 25K regulations.
I integrated this model with microarray data to find the “most probable” regulatory network using a parsimony criterium.
Once we get a map of the regulatory interactions, we can use it how the cell will evolve. Considering only the vector of RNA concentrations, we have the status \[C_t = (c_1,\ldots,c_n)_{t}\] Then the regulatory network defines \(F\) such that \[C_{t+1} = F(C_{t})\] Finding \(F\) is an open problem.
Broadly speaking, there are three kinds of proteins
We predict which genes code enzymes
Each enzyme catalyzes a reaction, with a known stoichiometry
Every reaction gives an equation
All equations plus boundary conditions give model to predict metabolite concentration
We can predict how the cell adapts to environmental changes
See: Flux Balance Analysis